Center for Human Identification, University of North Texas Health Science Center, 3500 Camp, Bowie Blvd., Fort Worth, TX 76107, USA.
Department of Microbiology, Immunology and Genetics, University of North Texas Health Science Center, 3500 Camp Bowie Blvd., Fort Worth, TX 76107, USA.
Genes (Basel). 2021 Jan 20;12(2):128. doi: 10.3390/genes12020128.
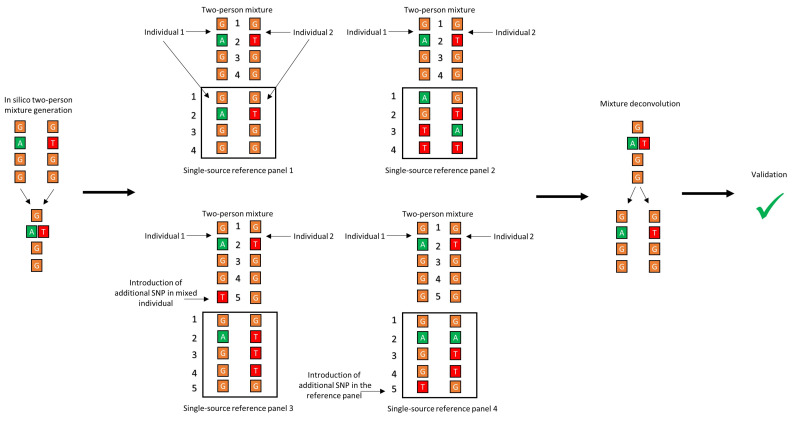
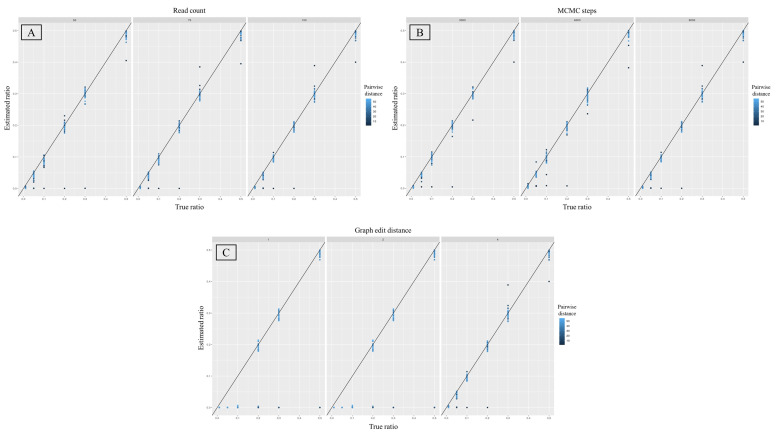
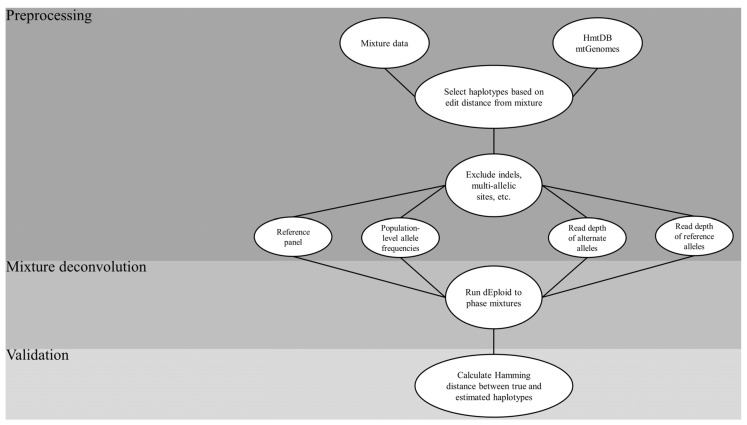
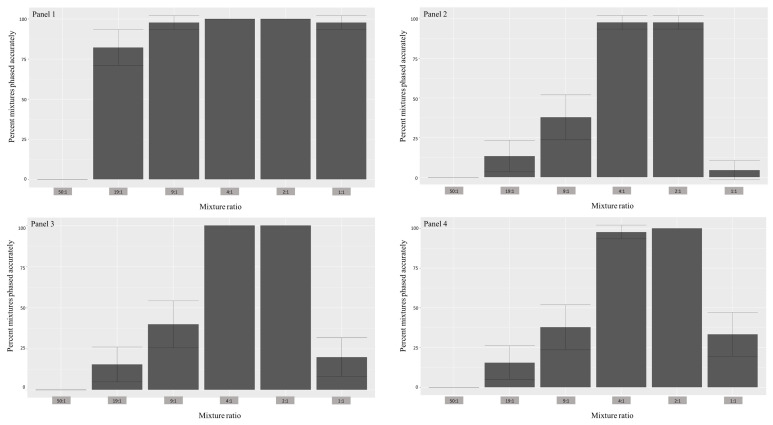
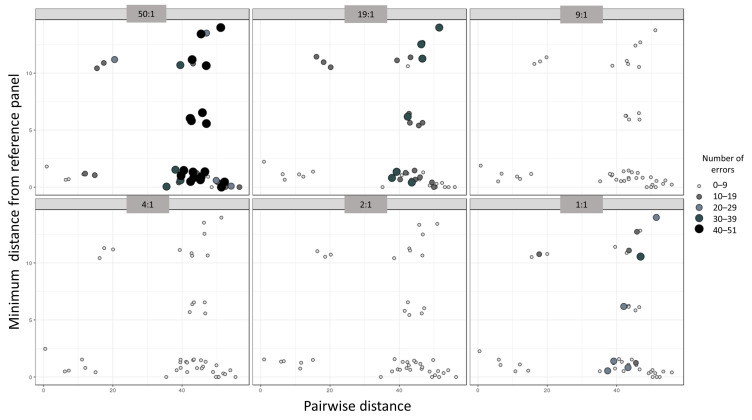
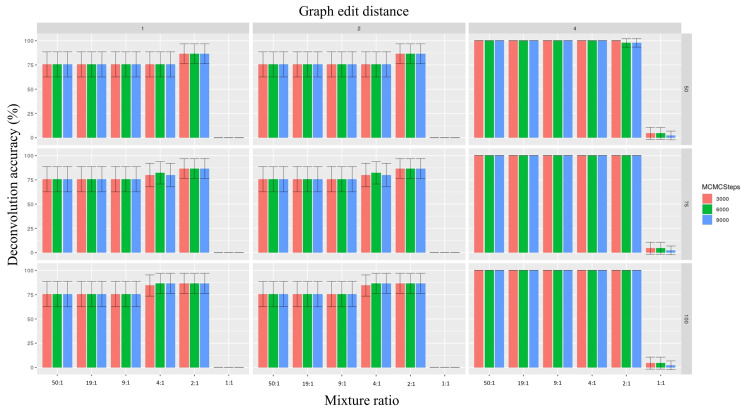
Despite the benefits of quantitative data generated by massively parallel sequencing, resolving mitotypes from mixtures occurring in certain ratios remains challenging. In this study, a bioinformatic mixture deconvolution method centered on population-based phasing was developed and validated. The method was first tested on 270 in silico two-person mixtures varying in mixture proportions. An assortment of external reference panels containing information on haplotypic variation (from similar and different haplogroups) was leveraged to assess the effect of panel composition on phasing accuracy. Building on these simulations, mitochondrial genomes from the Human Mitochondrial DataBase were sourced to populate the panels and key parameter values were identified by deconvolving an additional 7290 in silico two-person mixtures. Finally, employing an optimized reference panel and phasing parameters, the approach was validated with in vitro two-person mixtures with differing proportions. Deconvolution was most accurate when the haplotypes in the mixture were similar to haplotypes present in the reference panel and when the mixture ratios were neither highly imbalanced nor subequal (e.g., 4:1). Overall, errors in haplotype estimation were largely bounded by the accuracy of the mixture's genotype results. The proposed framework is the first available approach that automates the reconstruction of complete individual mitotypes from mixtures, even in ratios that have traditionally been considered problematic.
尽管大规模平行测序产生的定量数据有很多好处,但从某些特定比例的混合物中解析出线粒体型仍然具有挑战性。在这项研究中,开发并验证了一种以基于群体定相为中心的生物信息学混合物解卷积方法。该方法首先在 270 个不同混合比例的虚拟二人混合物上进行了测试。利用包含单倍型变异信息的各种外部参考面板(来自相似和不同的单倍型群)来评估面板组成对定相准确性的影响。基于这些模拟,从人类线粒体数据库中获取线粒体基因组以填充面板,并通过解卷积另外 7290 个虚拟二人混合物来确定关键参数值。最后,使用优化的参考面板和定相参数,该方法通过具有不同比例的体外二人混合物进行了验证。当混合物中的单倍型与参考面板中存在的单倍型相似,并且混合物比例既不过分不平衡也不相等(例如 4:1)时,解卷积的准确性最高。总体而言,单倍型估计的误差在很大程度上受到混合物基因型结果准确性的限制。该框架是第一个可自动从混合物中重建完整个体线粒体型的方法,即使在传统上被认为有问题的比例下也是如此。