Department of Computational Biology, University of Lausanne, Génopode, 1015, Lausanne, Switzerland.
Swiss Institute of Bioinformatics (SIB), University of Lausanne, Quartier Sorge - Batiment Amphipole, 1015, Lausanne, Switzerland.
Nat Commun. 2019 Nov 28;10(1):5436. doi: 10.1038/s41467-019-13225-y.
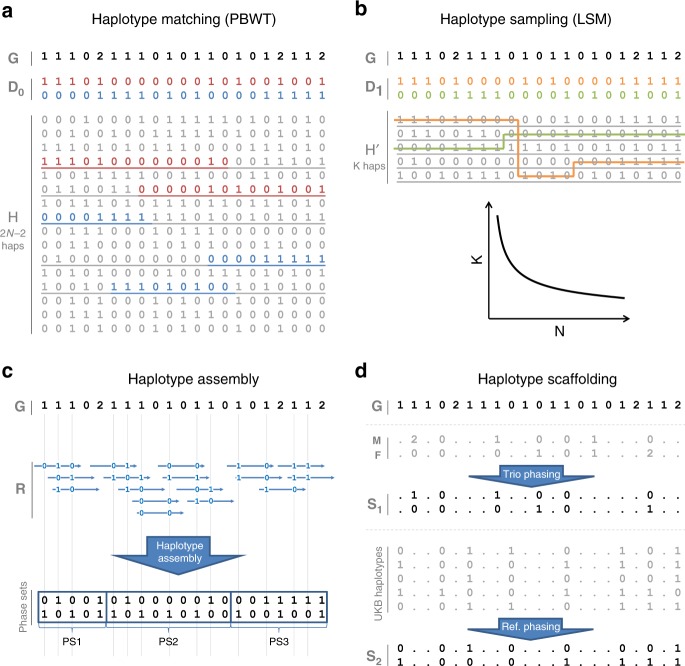
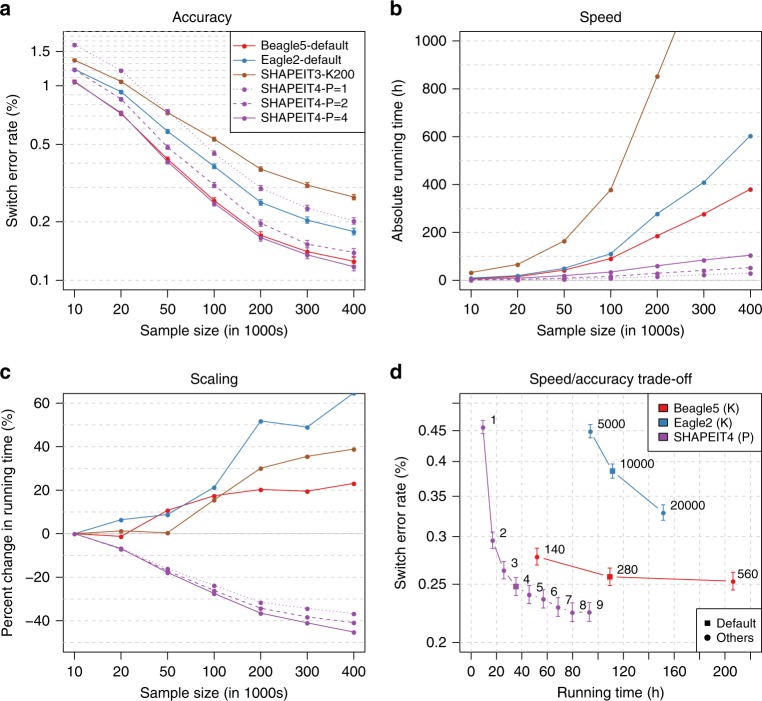
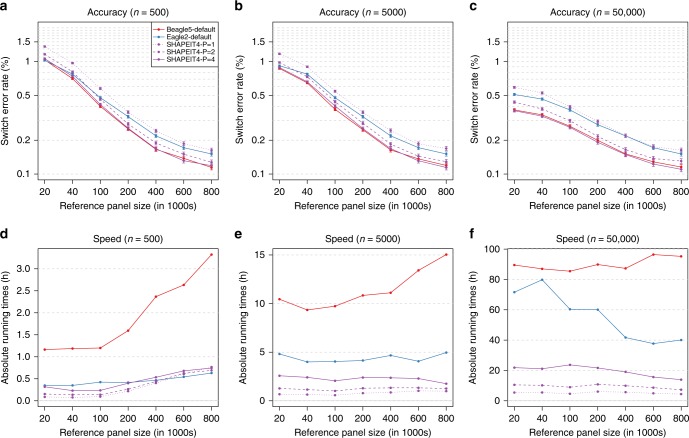
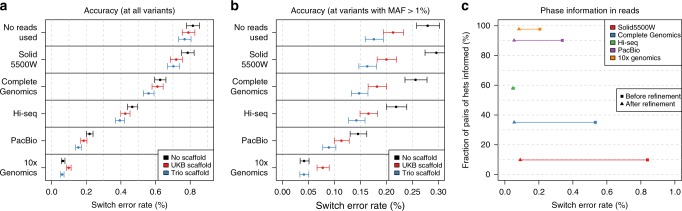
The number of human genomes being genotyped or sequenced increases exponentially and efficient haplotype estimation methods able to handle this amount of data are now required. Here we present a method, SHAPEIT4, which substantially improves upon other methods to process large genotype and high coverage sequencing datasets. It notably exhibits sub-linear running times with sample size, provides highly accurate haplotypes and allows integrating external phasing information such as large reference panels of haplotypes, collections of pre-phased variants and long sequencing reads. We provide SHAPEIT4 in an open source format and demonstrate its performance in terms of accuracy and running times on two gold standard datasets: the UK Biobank data and the Genome In A Bottle.
人类基因组的基因分型或测序数量呈指数级增长,现在需要能够处理这种数量数据的高效单倍型估计方法。在这里,我们提出了一种方法 SHAPEIT4,它极大地改进了其他方法来处理大型基因型和高覆盖测序数据集。它显著表现出与样本大小呈次线性的运行时间,提供高度准确的单倍型,并允许整合外部相位信息,如大型参考单倍型面板、预定相变体集合和长测序reads。我们以开源格式提供 SHAPEIT4,并在两个黄金标准数据集(英国生物银行数据和基因组瓶)上展示其在准确性和运行时间方面的性能。