Edwards J E, Shetty S A, van den Berg P, Burden F, van Doorn D A, Pellikaan W F, Dijkstra J, Smidt H
Laboratory of Microbiology, Wageningen University & Research, Wageningen, 6708 WE, Netherlands.
The Donkey Sanctuary, Sidmouth, Devon, EX10 ONU, UK.
Anim Microbiome. 2020 Feb 12;2(1):6. doi: 10.1186/s42523-020-0023-1.
Equine gut microbiology studies to date have primarily focused on horses and ponies, which represent only one of the eight extant equine species. This is despite asses and mules comprising almost half of the world's domesticated equines, and donkeys being superior to horses/ponies in their ability to degrade dietary fiber. Limited attention has also been given to commensal anaerobic fungi and archaea even though anaerobic fungi are potent fiber degrading organisms, the activity of which is enhanced by methanogenic archaea. Therefore, the objective of this study was to broaden the current knowledge of bacterial, anaerobic fungal and archaeal diversity of the equine fecal microbiota to multiple species of equines. Core taxa shared by all the equine fecal samples (n = 70) were determined and an overview given of the microbiota across different equine types (horse, donkey, horse × donkey and zebra).
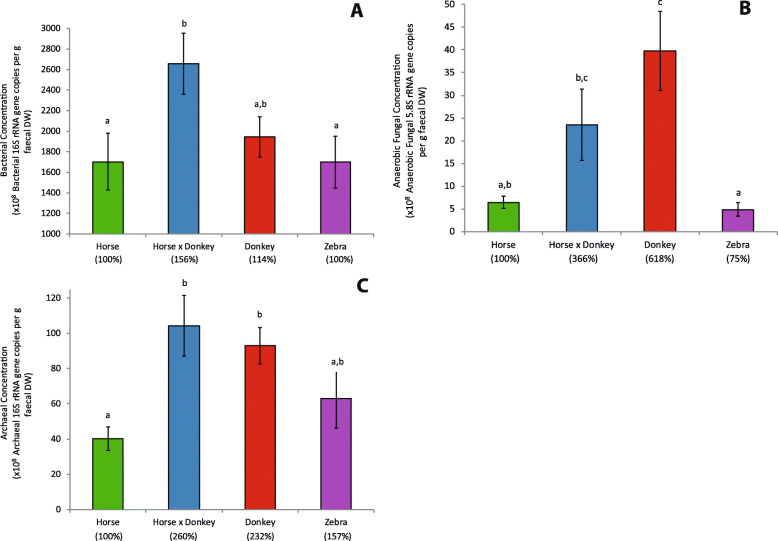
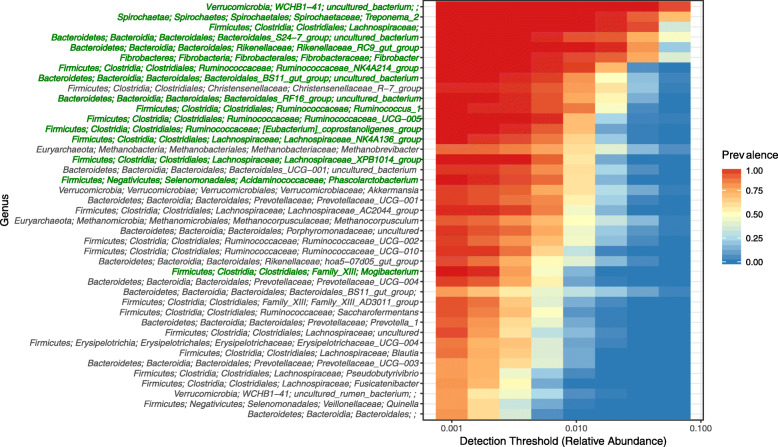
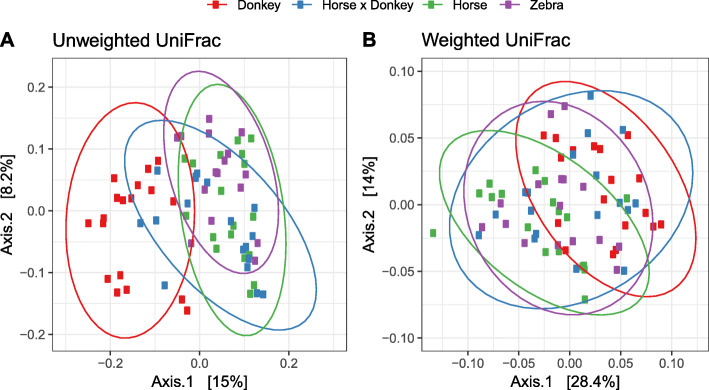
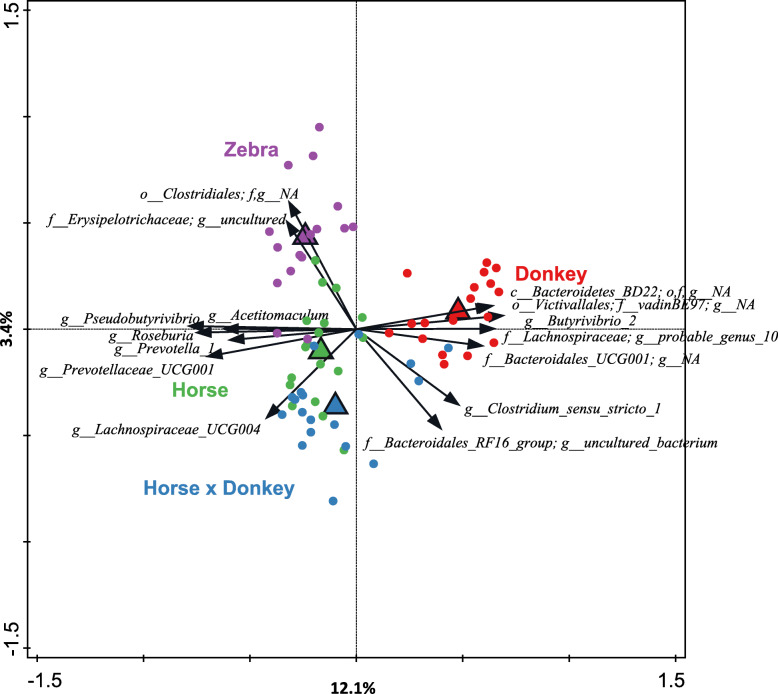
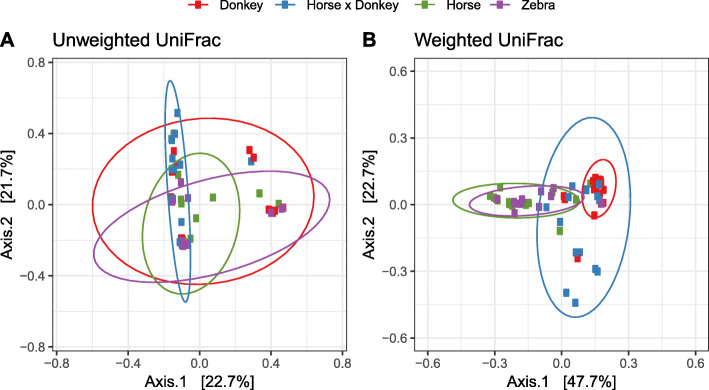
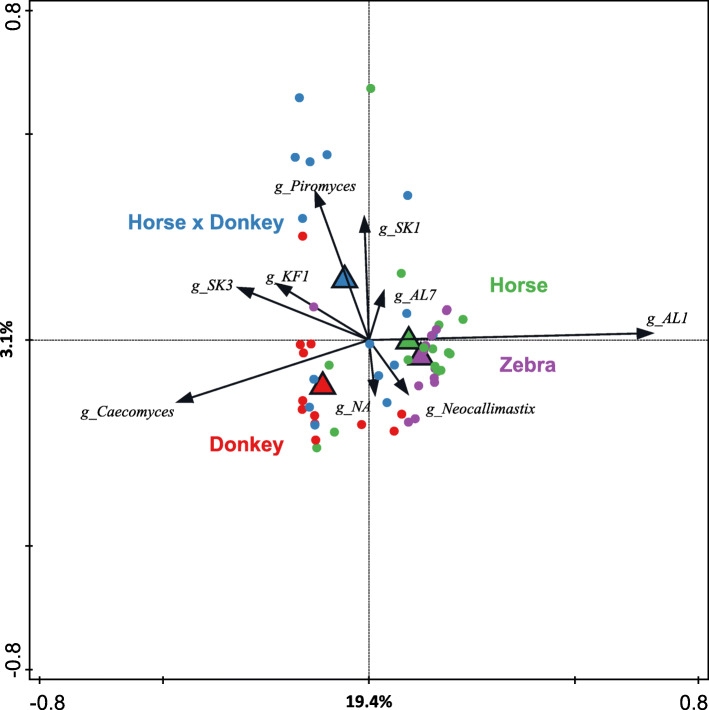
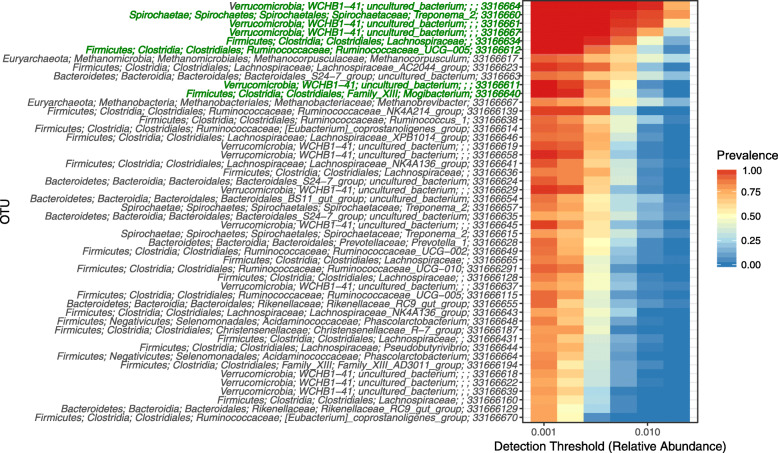
Equine type was associated with differences in both fecal microbial concentrations and community composition. Donkey was generally most distinct from the other equine types, with horse and zebra not differing. Despite this, a common bacterial core of eight OTUs (out of 2070) and 16 genus level groupings (out of 231) was found in all the fecal samples. This bacterial core represented a much larger proportion of the equine fecal microbiota than previously reported, primarily due to the detection of predominant core taxa belonging to the phyla Kiritimatiellaeota (formerly Verrucomicrobia subdivision 5) and Spirochaetes. The majority of the core bacterial taxa lack cultured representation. Archaea and anaerobic fungi were present in all animals, however, no core taxon was detected for either despite several taxa being prevalent and predominant.
Whilst differences were observed between equine types, a core fecal microbiota existed across all the equines. This core was composed primarily of a few predominant bacterial taxa, the majority of which are novel and lack cultured representation. The lack of microbial cultures representing the predominant taxa needs to be addressed, as their availability is essential to gain fundamental knowledge of the microbial functions that underpin the equine hindgut ecosystem.
迄今为止,马属动物肠道微生物学研究主要集中于马和矮种马,而它们只是现存八个马属物种中的一种。尽管驴和骡占全球家养马属动物的近一半,且驴在降解膳食纤维的能力上优于马和矮种马,但研究仍然有限。即使厌氧真菌是强效的纤维降解生物,且其活性会因产甲烷古菌而增强,共生厌氧真菌和古菌也同样受到较少关注。因此,本研究的目的是拓宽当前对多种马属动物粪便微生物群中细菌、厌氧真菌和古菌多样性的认识。确定了所有马属动物粪便样本(n = 70)共有的核心分类群,并概述了不同马属动物类型(马、驴、马×驴和斑马)的微生物群。
马属动物类型与粪便微生物浓度和群落组成的差异有关。驴通常与其他马属动物类型差异最大,马和斑马之间没有差异。尽管如此,在所有粪便样本中发现了一个由8个OTU(共2070个)和16个属水平分类群(共231个)组成的常见细菌核心。这个细菌核心在马属动物粪便微生物群中所占比例比之前报道的要大得多,这主要是由于检测到了属于基里马蒂埃洛菌门(原疣微菌门第5亚群)和螺旋体门的主要核心分类群。大多数核心细菌分类群缺乏培养代表菌株。所有动物体内均存在古菌和厌氧真菌,然而,尽管有几个分类群普遍且占主导地位,但未检测到它们的核心分类群。
虽然在马属动物类型之间观察到了差异,但所有马属动物都存在核心粪便微生物群。这个核心主要由一些占主导地位的细菌分类群组成,其中大多数是新发现的且缺乏培养代表菌株。需要解决缺乏代表主要分类群的微生物培养物的问题,因为它们对于了解支撑马属动物后肠生态系统的微生物功能的基础知识至关重要。