Department of Bioengineering and Barnett Institute, Northeastern University, Boston, MA, 02115, USA.
Present Address: Department of Biochemistry, Centre for Proteome Research, University of Liverpool, Liverpool, L69 7ZB, UK.
Genome Biol. 2021 Jan 27;22(1):50. doi: 10.1186/s13059-021-02267-5.
Macrophages are innate immune cells with diverse functional and molecular phenotypes. This diversity is largely unexplored at the level of single-cell proteomes because of the limitations of quantitative single-cell protein analysis.
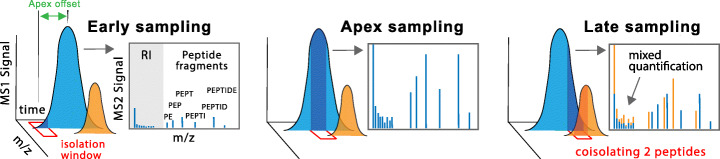
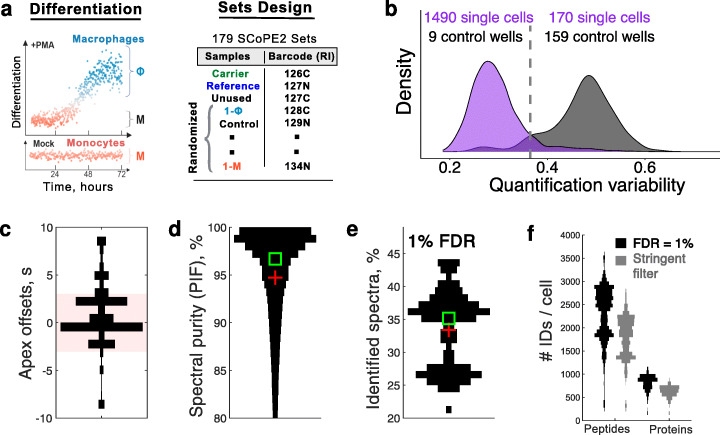
To overcome this limitation, we develop SCoPE2, which substantially increases quantitative accuracy and throughput while lowering cost and hands-on time by introducing automated and miniaturized sample preparation. These advances enable us to analyze the emergence of cellular heterogeneity as homogeneous monocytes differentiate into macrophage-like cells in the absence of polarizing cytokines. SCoPE2 quantifies over 3042 proteins in 1490 single monocytes and macrophages in 10 days of instrument time, and the quantified proteins allow us to discern single cells by cell type. Furthermore, the data uncover a continuous gradient of proteome states for the macrophages, suggesting that macrophage heterogeneity may emerge in the absence of polarizing cytokines. Parallel measurements of transcripts by 10× Genomics suggest that our measurements sample 20-fold more protein copies than RNA copies per gene, and thus, SCoPE2 supports quantification with improved count statistics. This allowed exploring regulatory interactions, such as interactions between the tumor suppressor p53, its transcript, and the transcripts of genes regulated by p53.
Even in a homogeneous environment, macrophage proteomes are heterogeneous. This heterogeneity correlates to the inflammatory axis of classically and alternatively activated macrophages. Our methodology lays the foundation for automated and quantitative single-cell analysis of proteins by mass spectrometry and demonstrates the potential for inferring transcriptional and post-transcriptional regulation from variability across single cells.
巨噬细胞是先天免疫细胞,具有多种功能和分子表型。由于定量单细胞蛋白质分析的局限性,这种多样性在单细胞蛋白质组学水平上很大程度上尚未得到探索。
为了克服这一限制,我们开发了 SCoPE2,它通过引入自动化和小型化的样品制备,在提高定量准确性和通量的同时,降低了成本和人工时间。这些进展使我们能够分析细胞异质性的出现,因为同质单核细胞在没有极化细胞因子的情况下分化为巨噬细胞样细胞。SCoPE2 在 10 天的仪器时间内定量分析了 1490 个单核细胞和巨噬细胞中的 3042 多种蛋白质,并且定量的蛋白质允许我们按细胞类型区分单细胞。此外,这些数据揭示了巨噬细胞中蛋白质组状态的连续梯度,表明在没有极化细胞因子的情况下,巨噬细胞的异质性可能会出现。通过 10× Genomics 平行测量转录本表明,我们的测量方法每基因的蛋白质拷贝数比 RNA 拷贝数多 20 倍,因此,SCoPE2 支持计数统计数据改善后的定量分析。这使得探索调控相互作用成为可能,例如肿瘤抑制因子 p53、其转录本和受 p53 调控的基因转录本之间的相互作用。
即使在同质环境中,巨噬细胞蛋白质组也是异质的。这种异质性与经典和替代激活的巨噬细胞的炎症轴相关。我们的方法为通过质谱对蛋白质进行自动化和定量单细胞分析奠定了基础,并展示了从单细胞间的变异性推断转录和转录后调控的潜力。