Amirkabir University of Technology, Tehran 346512, Iran.
AI-enabled Processes (AIP) Research Centre, Health Data Analytics Program, Macquarie University, Sydney, NSW 2109, Australia.
Genes (Basel). 2021 Jan 27;12(2):186. doi: 10.3390/genes12020186.
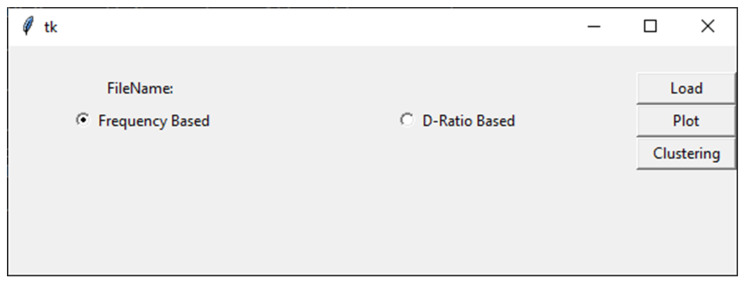
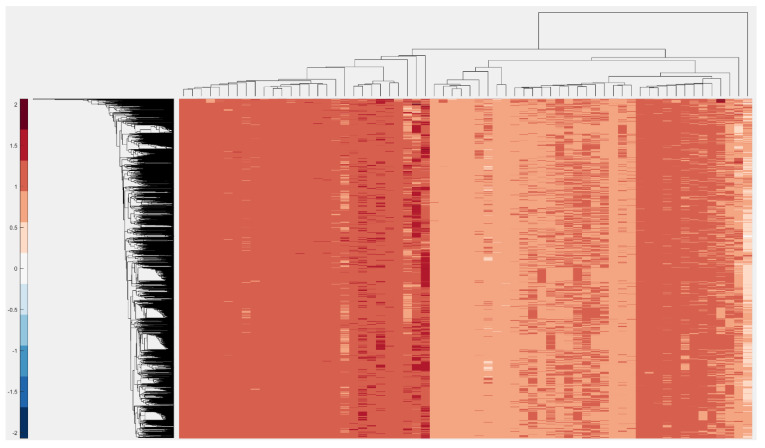
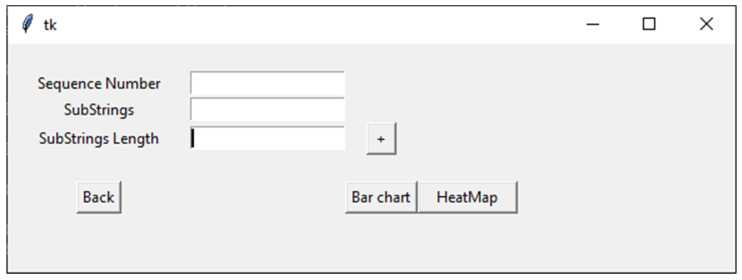
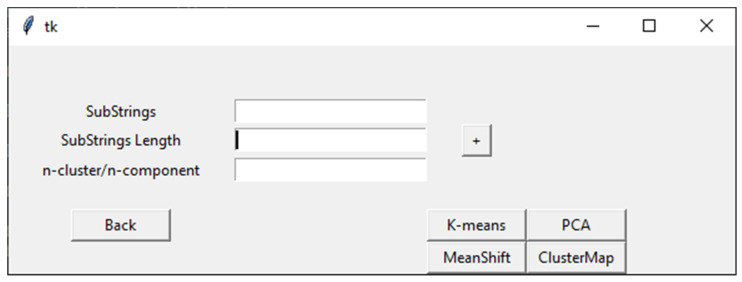
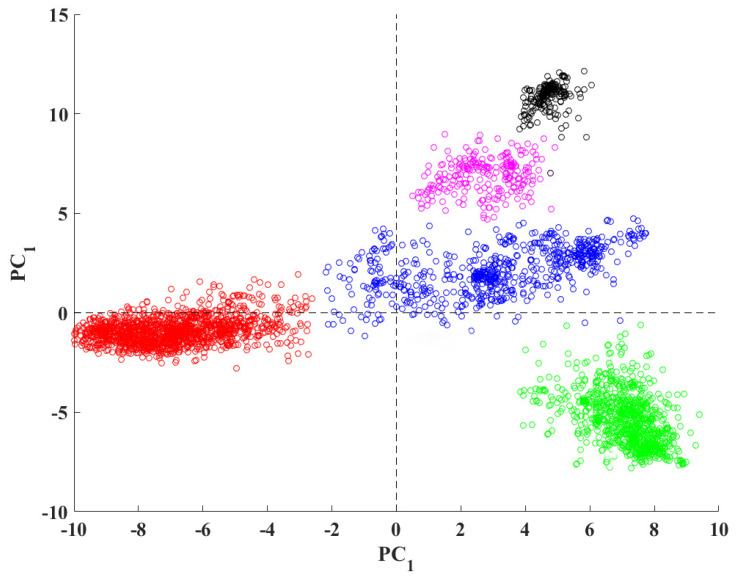
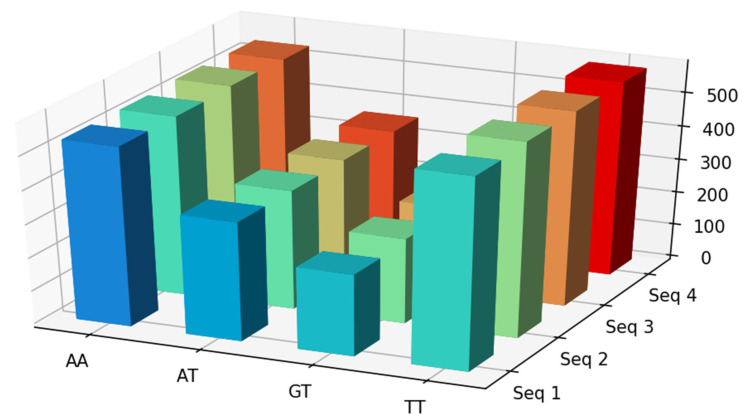
Bioinformatics and computational biology have significantly contributed to the generation of vast and important knowledge that can lead to great improvements and advancements in biology and its related fields. Over the past three decades, a wide range of tools and methods have been developed and proposed to enhance performance, diagnosis, and throughput while maintaining feasibility and convenience for users. Here, we propose a new user-friendly comprehensive tool called VIRMOTIF to analyze DNA sequences. VIRMOTIF brings different tools together as one package so that users can perform their analysis as a whole and in one place. VIRMOTIF is able to complete different tasks, including computing the number or probability of motifs appearing in DNA sequences, visualizing data using the matplotlib and heatmap libraries, and clustering data using four different methods, namely K-means, PCA, Mean Shift, and ClusterMap. VIRMOTIF is the only tool with the ability to analyze genomic motifs based on their frequency and representation (D-ratio) in a virus genome.
生物信息学和计算生物学为生成大量重要知识做出了重大贡献,这些知识可以为生物学及其相关领域带来巨大的改进和进步。在过去的三十年中,已经开发和提出了广泛的工具和方法,以提高性能、诊断和吞吐量,同时保持用户的可行性和便利性。在这里,我们提出了一个名为 VIRMOTIF 的新的用户友好型综合工具,用于分析 DNA 序列。VIRMOTIF 将不同的工具集成在一起作为一个包,以便用户可以整体地在一个地方进行分析。VIRMOTIF 能够完成不同的任务,包括计算 DNA 序列中出现的基序的数量或概率,使用 matplotlib 和 heatmap 库可视化数据,以及使用四种不同的方法(K-means、PCA、Mean Shift 和 ClusterMap)对数据进行聚类。VIRMOTIF 是唯一能够根据病毒基因组中基序的频率和表示(D-比)分析基因组基序的工具。