Banfi Felipe Finger, Krombauer Gabriela Camila, da Fonseca Amanda Luisa, Nunes Renata Rachide, Andrade Silmara Nunes, de Rezende Millena Alves, Chaves Mariana Helena, Monção Evaldo Dos Santos, Taranto Alex Guterres, Rodrigues Domingos de Jesus, Vieira Gerardo Magela, de Castro Whocely Victor, Varotti Fernando de Pilla, Sanchez Bruno Antonio Marinho
Laboratory of Immunopathology and Tropical Diseases, Health Education and Research Center (NUPADS), Institute of Health Sciences, Federal University of Mato Grosso, Sinop, MT, Brazil.
Research Center on Biological Chemistry (NQBio), Federal University of São João Del Rei, Divinópolis, MG, Brazil.
J Venom Anim Toxins Incl Trop Dis. 2021 Jan 8;27:e20200073. doi: 10.1590/1678-9199-JVATITD-2020-0073. eCollection 2021.
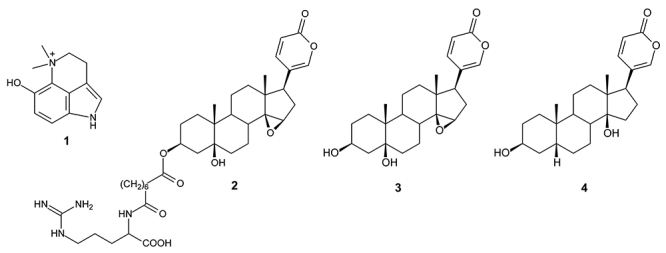
The resistance against antimalarial drugs represents a global challenge in the fight and control of malaria. The Brazilian biodiversity can be an important tool for research and development of new medicinal products. In this context, toxinology is a multidisciplinary approach on the development of new drugs, including the isolation, purification, and evaluation of the pharmacological activities of natural toxins. The present study aimed to evaluate the cytotoxicity, as well as the antimalarial activity and of four compounds isolated from venom as potential oral drug prototypes.
Four compounds were challenged against 35 target proteins from and screened to evaluate their physicochemical properties using docking assay in Brazilian Malaria Molecular Targets (BraMMT) software and assay in OCTOPUS® software. The antimalarial activity of the compounds against the 3D7 clones were assessed using the SYBR Green I based assay (IC). For the cytotoxic tests, the LD was determined in human pulmonary fibroblast cell line using the [3(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] (MTT) assay.
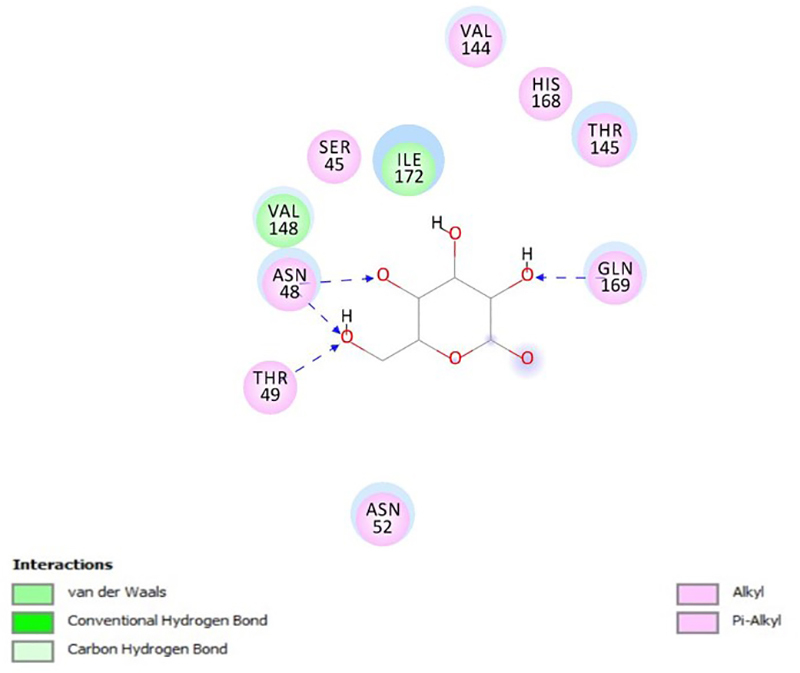
All compounds presented a ligand-receptor interaction with ten -related protein targets, as well as antimalarial activity against chloroquine resistant strain (IC = 3.44 μM to 19.11 μM). Three of them (dehydrobufotenine, marinobufagin, and bufalin) showed adequate conditions for oral drug prototypes, with satisfactory prediction of absorption, permeability, and absence of toxicity. In the cell viability assay, only dehydrobufotenin was selective for the parasite.
Dehydrobufotenin revealed to be a potential oral drug prototype presenting adequate antimalarial activity and absence of cytotoxicity, therefore should be subjected to further studies.
抗疟药物耐药性是疟疾防治中的一项全球性挑战。巴西的生物多样性可能成为新药研发的重要工具。在此背景下,毒素学是一种开发新药的多学科方法,包括天然毒素的分离、纯化及药理活性评估。本研究旨在评估从毒液中分离出的四种化合物作为潜在口服药物原型的细胞毒性、抗疟活性。
针对来自巴西疟疾分子靶点(BraMMT)软件中35种靶蛋白对四种化合物进行测试,并使用OCTOPUS®软件中的对接分析来筛选评估其理化性质。采用基于SYBR Green I的检测方法(IC)评估这些化合物对3D7克隆的抗疟活性。对于细胞毒性测试,使用[3-(4,5-二甲基噻唑-2-基)-2,5-二苯基四氮唑溴盐](MTT)检测法在人肺成纤维细胞系中测定半数致死量(LD)。
所有化合物均与十种疟原虫相关蛋白靶点呈现配体-受体相互作用,并且对氯喹耐药菌株具有抗疟活性(IC = 3.44 μM至19.11 μM)。其中三种化合物(去氢蟾蜍色胺、海蟾蜍精和蟾毒灵)显示出作为口服药物原型具备合适的条件,在吸收、渗透性预测方面令人满意且无毒性。在细胞活力测定中,只有去氢蟾蜍色胺对疟原虫具有选择性。
去氢蟾蜍色胺显示为一种潜在的口服药物原型,具有足够的抗疟活性且无细胞毒性,因此应进行进一步研究。