Enqi Wu, Jingzhu Song, Lingpeng Pei, Yaqin Ling
Key Laboratory of Ethnomedicine (Minzu University of China), Ministry of Education, Beijing, China.
Front Cell Infect Microbiol. 2021 Jan 14;10:581974. doi: 10.3389/fcimb.2020.581974. eCollection 2020.
The study aimed to identify the effects of modeling procedures on bacterial communities and to investigate whether different modeling procedures lead to consistent patterns of gut microbiome compositions.
Two irritable bowel syndrome (IBS) rat models maternal separation (MS) alone and multiple-early-adversity modeling (MAM) were established and the gut microbiome were analyzed using 16S-rRNA-based high-throughput sequencing methods.
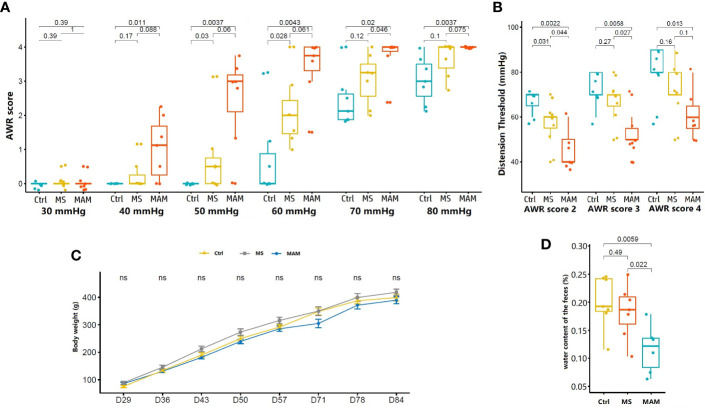
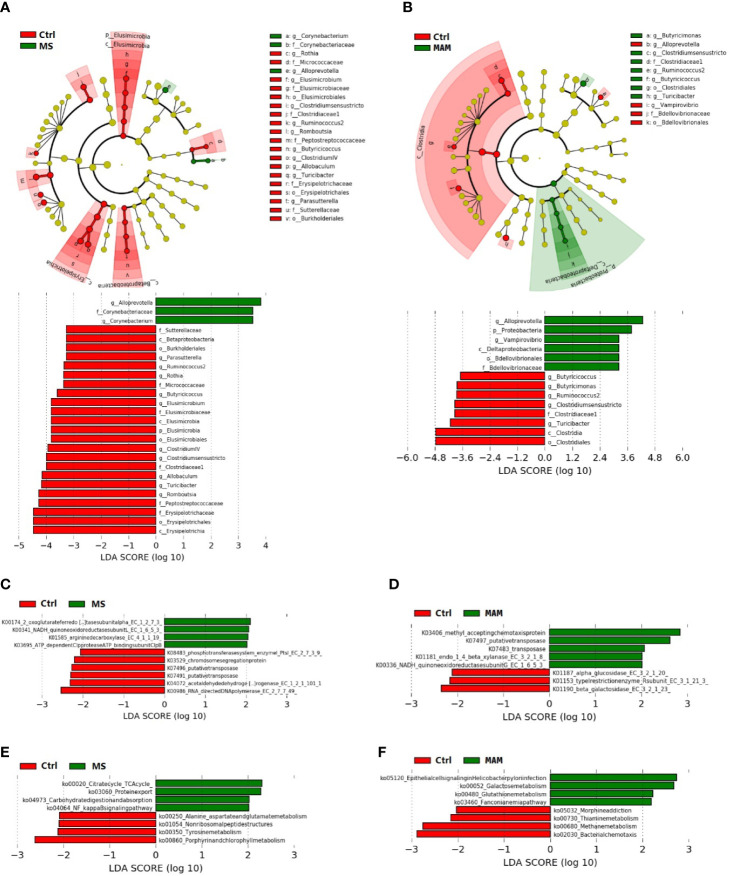
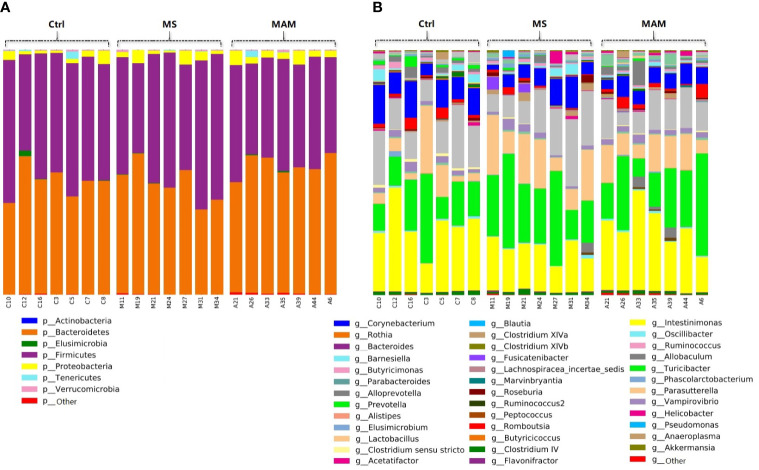
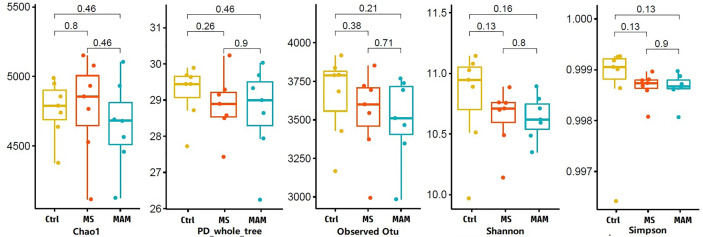
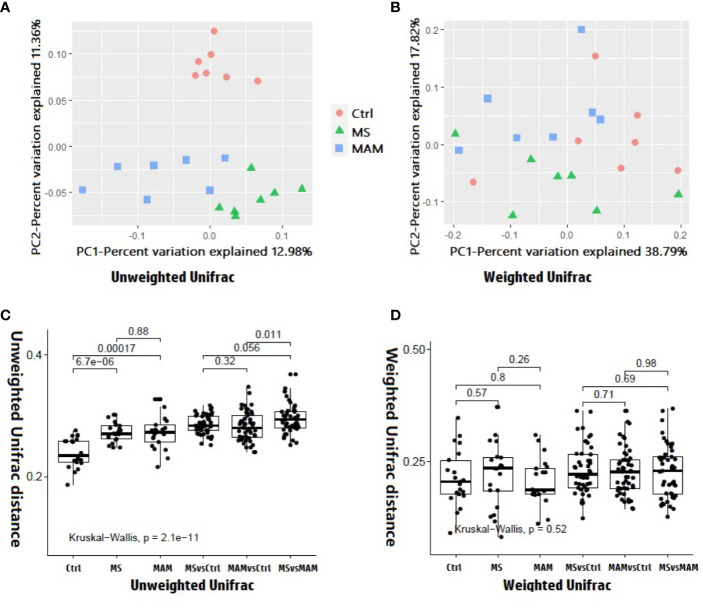
Rats from both models exhibited visceral hypersensitivity and the two model groups exhibited differences in the extent of visceral sensitivity and fecal water content. The microbial community structure of the two models exhibited significant differences compared to the controls, while the two model groups also exhibited significant differences between them. Furthermore, microbial community functional predictions suggested that the two models exhibited different abundances of metabolisms and pathways. Several common and distinct characteristic differences were also observed between the two model groups. were more abundant in both model groups, while , , , and along with the family it belongs to were less abundant relative to controls. In addition, the abundance of , , , , , , , and their related taxa were specifically associated with MS group, whereas and along with its related taxa were specifically associated with MAM group. Among those, , and were found to partially mediate early adversity exposure-induced visceral hypersensitivity.
Our results highlight the importance in evaluating gut microbiota characteristics in IBS research while also systematically considering potential modeling procedural differences. The microbial compositional/functional differences identified in this study were suggestive to further investigation of mechanisms of early adversity induced IBS.
本研究旨在确定建模程序对细菌群落的影响,并调查不同的建模程序是否会导致肠道微生物群组成的一致模式。
建立了两种肠易激综合征(IBS)大鼠模型,即单独的母婴分离(MS)模型和多重早期逆境建模(MAM)模型,并使用基于16S - rRNA的高通量测序方法分析肠道微生物群。
两个模型的大鼠均表现出内脏超敏反应,且两个模型组在内脏敏感性程度和粪便含水量方面存在差异。与对照组相比,两种模型的微生物群落结构均表现出显著差异,而两个模型组之间也表现出显著差异。此外,微生物群落功能预测表明,两种模型在代谢和途径的丰度上存在差异。在两个模型组之间还观察到了一些共同和独特的特征差异。在两个模型组中均更为丰富,而相对于对照组, 、 、 和 及其所属家族的丰度较低。此外, 、 、 、 、 、 、 和它们相关分类群的丰度与MS组特异性相关,而 及其相关分类群与MAM组特异性相关。其中, 、 和 被发现部分介导了早期逆境暴露诱导的内脏超敏反应。
我们的结果强调了在IBS研究中评估肠道微生物群特征的重要性,同时也需要系统地考虑潜在的建模程序差异。本研究中确定的微生物组成/功能差异提示需要进一步研究早期逆境诱导IBS的机制。