Jairajpuri Deeba Shamim, Hussain Afzal, Nasreen Khalida, Mohammad Taj, Anjum Farah, Tabish Rehman Md, Mustafa Hasan Gulam, Alajmi Mohamed F, Imtaiyaz Hassan Md
Department of Medical Biochemistry, College of Medicine and Medical Sciences, Arabian Gulf University, P.O. Box 22971, Manama, Bahrain.
Department of Pharmacognosy, College of Pharmacy, King Saud University, Riyadh 11451, Saudi Arabia.
Saudi J Biol Sci. 2021 Apr;28(4):2423-2431. doi: 10.1016/j.sjbs.2021.01.040. Epub 2021 Jan 27.
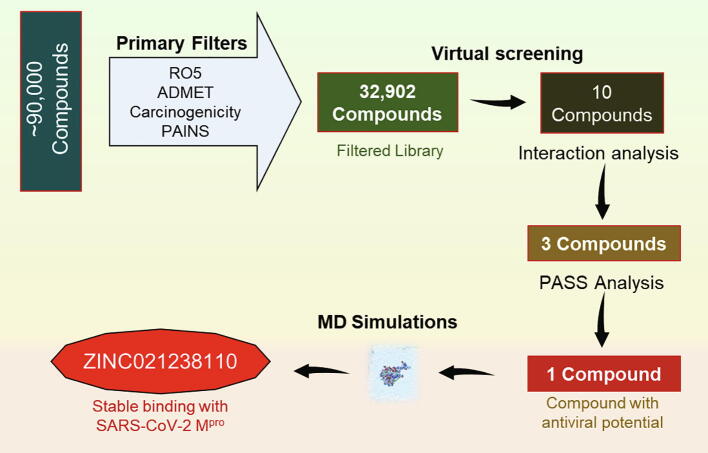
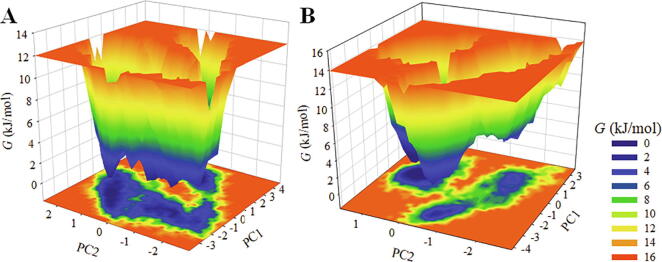
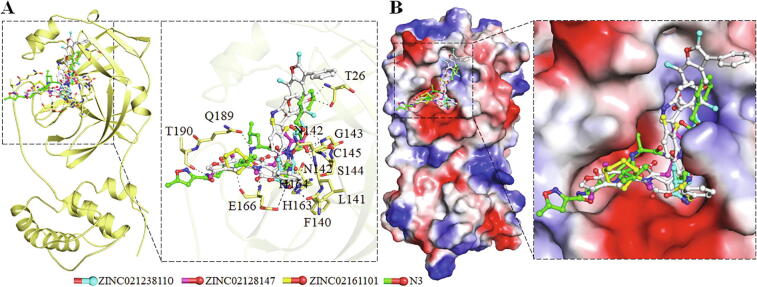
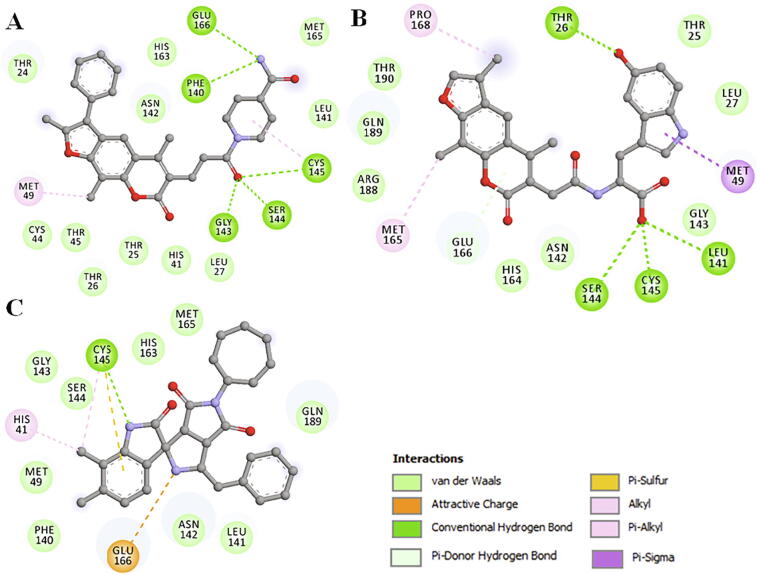
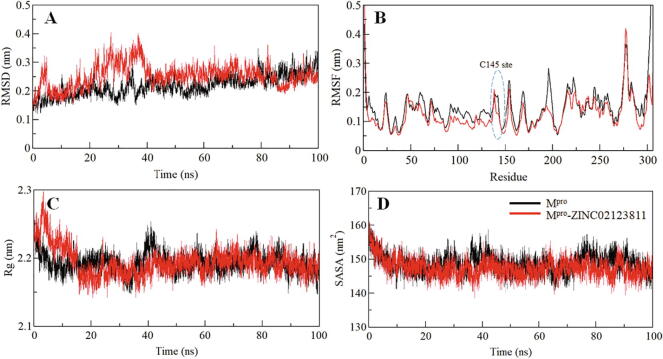
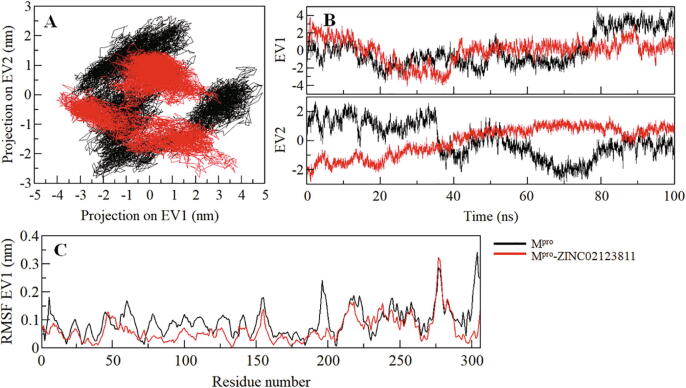
Coronavirus disease 2019 (COVID-19) has emerged from China and globally affected the entire population through the human-to-human transmission of a newly emerged virus called severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). The genome of SARS-CoV-2 encodes several proteins that are essential for multiplication and pathogenesis. The main protease (M or 3CL) of SARS-CoV-2 plays a central role in its pathogenesis and thus is considered as an attractive drug target for the drug design and development of small-molecule inhibitors. We have employed an extensive structure-based high-throughput virtual screening to discover potential natural compounds from the ZINC database which could inhibit the M of SARS-CoV-2. Initially, the hits were selected on the basis of their physicochemical and drug-like properties. Subsequently, the PAINS filter, estimation of binding affinities using molecular docking, and interaction analyses were performed to find safe and potential inhibitors of SARS-CoV-2 M. We have identified ZINC02123811 (1-(3-(2,5,9-trimethyl-7-oxo-3-phenyl-7H-furo[3,2-g]chromen-6-yl)propanoyl)piperidine-4-carboxamide), a natural compound bearing appreciable affinity, efficiency, and specificity towards the binding pocket of SARS-CoV-2 M. The identified compound showed a set of drug-like properties and preferentially binds to the active site of SARS-CoV-2 M. All-atom molecular dynamics (MD) simulations were performed to evaluate the conformational dynamics, stability and interaction mechanism of M with ZINC02123811. MD simulation results indicated that M with ZINC02123811 forms a stable complex throughout the trajectory of 100 ns. These findings suggest that ZINC02123811 may be further exploited as a promising scaffold for the development of potential inhibitors of SARS-CoV-2 M to address COVID-19.
2019年冠状病毒病(COVID-19)起源于中国,并通过一种新出现的病毒——严重急性呼吸综合征冠状病毒2(SARS-CoV-2)的人际传播在全球范围内影响了整个人口。SARS-CoV-2的基因组编码了几种对病毒复制和发病机制至关重要的蛋白质。SARS-CoV-2的主要蛋白酶(M或3CL)在其发病机制中起着核心作用,因此被认为是小分子抑制剂药物设计和开发的有吸引力的药物靶点。我们采用了基于结构的广泛高通量虚拟筛选,从ZINC数据库中发现可能抑制SARS-CoV-2 M的潜在天然化合物。最初,根据其物理化学和类药性质选择命中的化合物。随后,进行PAINS筛选、使用分子对接估计结合亲和力以及相互作用分析,以寻找SARS-CoV-2 M的安全且潜在的抑制剂。我们鉴定出了ZINC02123811(1-(3-(2,5,9-三甲基-7-氧代-3-苯基-7H-呋喃并[3,2-g]色烯-6-基)丙酰基)哌啶-4-甲酰胺),这是一种对SARS-CoV-2 M的结合口袋具有可观亲和力、效率和特异性的天然化合物。所鉴定的化合物表现出一系列类药性质,并优先结合到SARS-CoV-2 M的活性位点。进行了全原子分子动力学(MD)模拟,以评估M与ZINC02123811的构象动力学、稳定性和相互作用机制。MD模拟结果表明,M与ZINC021下3811在100纳秒的整个轨迹中形成了稳定的复合物。这些发现表明,ZINC02123811可能被进一步开发为一种有前景的支架,用于开发SARS-CoV-2 M的潜在抑制剂以应对COVID-19。