Department of Civil and Environmental Engineering, University of Michigan, Ann Arbor, Michigan, USA.
Ecology and Evolutionary Biology, University of Michigan, Ann Arbor, Michigan, USA.
mBio. 2021 Feb 2;12(1):e03173-20. doi: 10.1128/mBio.03173-20.
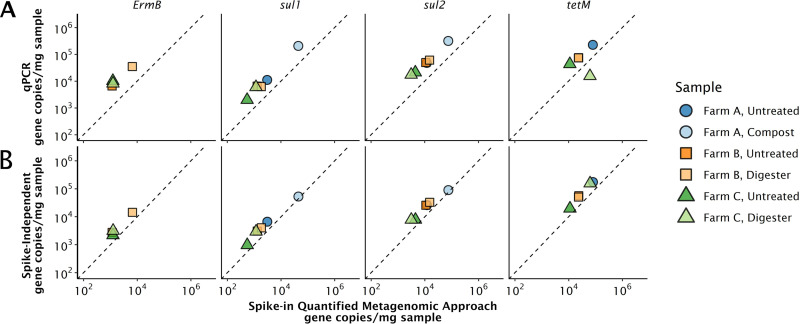
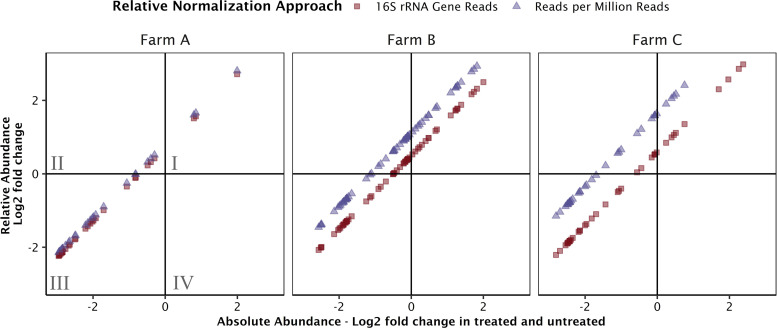
We demonstrate that an assembly-independent and spike-in facilitated metagenomic quantification approach can be used to screen and quantify over 2,000 genes simultaneously, while delivering absolute gene concentrations comparable to those for quantitative PCR (qPCR). DNA extracted from dairy manure slurry, digestate, and compost was spiked with genomic DNA from a marine bacterium and sequenced using the Illumina HiSeq4000. We compared gene copy concentrations, in gene copies per mass of sample, of five antimicrobial resistance genes (ARGs) generated with (i) our quantitative metagenomic approach, (ii) targeted qPCR, and (iii) a hybrid quantification approach involving metagenomics and qPCR-based 16S rRNA gene quantification. Although qPCR achieved lower quantification limits, the metagenomic method avoided biases caused by primer specificity inherent to qPCR-based methods and was able to detect orders of magnitude more genes than is possible with qPCR assays. We used the approach to simultaneously quantify ARGs in the Comprehensive Antimicrobial Resistance Database (CARD). We observed that the total abundance of tetracycline resistance genes was consistent across different stages of manure treatment on three farms, but different samples were dominated by different tetracycline resistance gene families. qPCR and metagenomics are central molecular techniques that have offered insights into biological processes for decades, from monitoring spatial and temporal gene dynamics to tracking ARGs or pathogens. Still needed is a tool that can quantify thousands of relevant genes in a sample as gene copies per sample mass or volume. We compare a quantitative metagenomic approach with traditional qPCR approaches in the quantification of ARG targets in dairy manure samples. By leveraging the benefits of nontargeted community genomics, we demonstrate high-throughput absolute gene quantification of all known ARG sequences in environmental samples.
我们证明了一种组装独立且 Spike-in 辅助的宏基因组定量方法可用于同时筛选和定量超过 2000 个基因,同时提供与定量 PCR(qPCR)相当的绝对基因浓度。从奶牛粪便泥浆、消化物和堆肥中提取的 DNA 用海洋细菌的基因组 DNA 进行 Spike-in,并使用 Illumina HiSeq4000 进行测序。我们比较了五种抗生素抗性基因(ARGs)的基因拷贝浓度,以样品质量的基因拷贝数表示,这些基因拷贝数是通过(i)我们的定量宏基因组方法、(ii)靶向 qPCR 和(iii)涉及宏基因组学和 qPCR 16S rRNA 基因定量的混合定量方法生成的。尽管 qPCR 实现了更低的定量极限,但宏基因组方法避免了 qPCR 方法固有的引物特异性引起的偏差,并且能够检测到 qPCR 检测的数量级更多的基因。我们使用该方法同时定量 CARD 中的 ARGs。我们观察到,在三个农场的不同粪便处理阶段,四环素抗性基因的总丰度是一致的,但不同的样本由不同的四环素抗性基因家族主导。qPCR 和宏基因组学是几十年来提供生物学过程见解的核心分子技术,从监测时空基因动态到跟踪 ARGs 或病原体。仍然需要一种能够以每个样品的基因拷贝数/样品质量或体积来定量样品中数千个相关基因的工具。我们在奶牛粪便样本中比较了定量宏基因组方法与传统 qPCR 方法在 ARG 靶标的定量。通过利用非靶向群落基因组学的优势,我们展示了环境样本中所有已知 ARG 序列的高通量绝对基因定量。