Institute of Integrative Biology, The University of Liverpool, Liverpool, UK.
Scientific Computing Department, The Hartree Centre, STFC Daresbury Laboratory, Warrington, UK.
Bioinformatics. 2018 Nov 1;34(21):3601-3608. doi: 10.1093/bioinformatics/bty387.
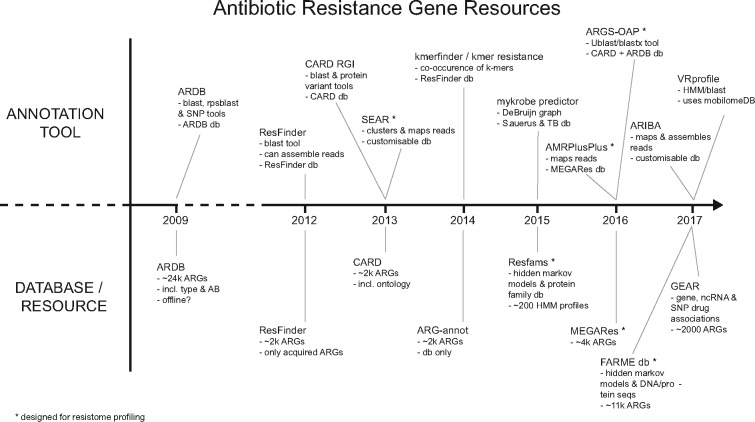
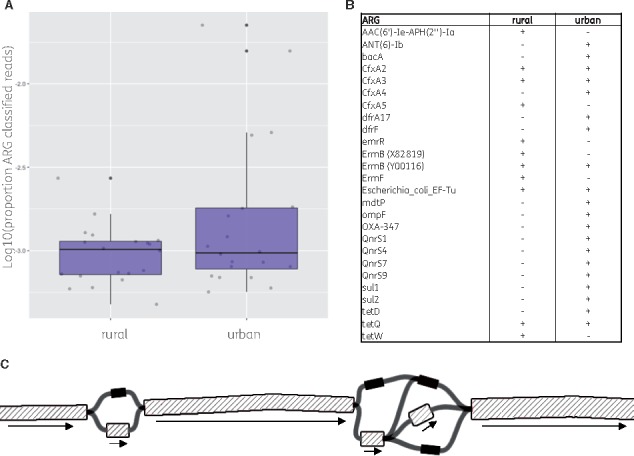
Antimicrobial resistance (AMR) remains a major threat to global health. Profiling the collective AMR genes within a metagenome (the 'resistome') facilitates greater understanding of AMR gene diversity and dynamics. In turn, this can allow for gene surveillance, individualized treatment of bacterial infections and more sustainable use of antimicrobials. However, resistome profiling can be complicated by high similarity between reference genes, as well as the sheer volume of sequencing data and the complexity of analysis workflows. We have developed an efficient and accurate method for resistome profiling that addresses these complications and improves upon currently available tools.
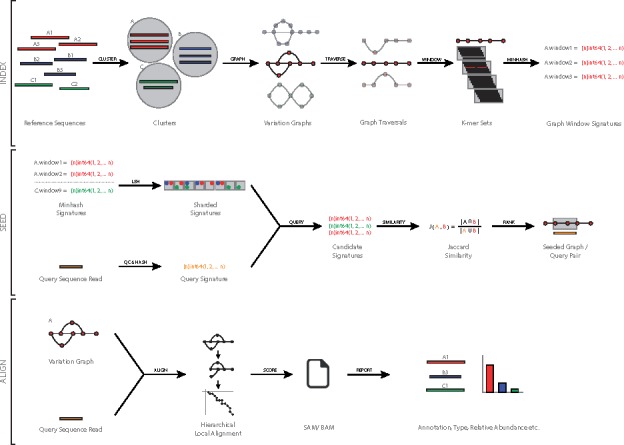
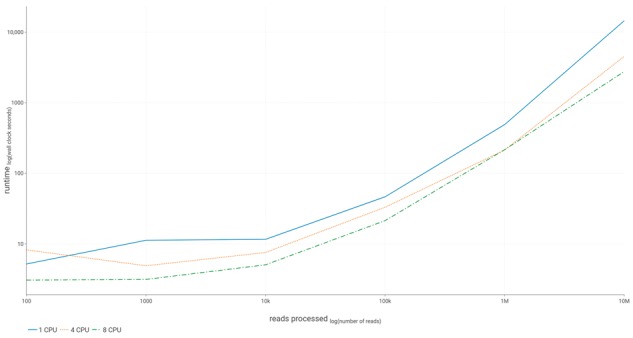
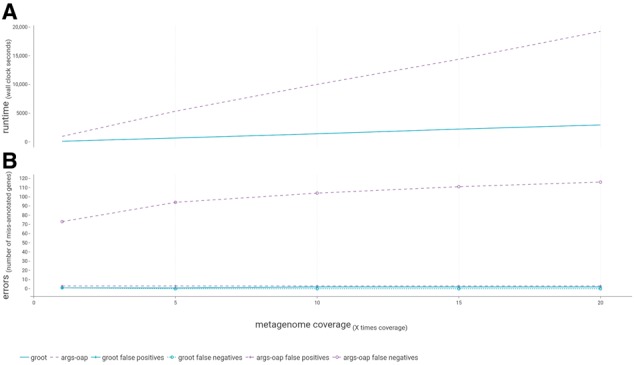
Our method combines a variation graph representation of gene sets with a locality-sensitive hashing Forest indexing scheme to allow for fast classification of metagenomic sequence reads using similarity-search queries. Subsequent hierarchical local alignment of classified reads against graph traversals enables accurate reconstruction of full-length gene sequences using a scoring scheme. We provide our implementation, graphing Resistance Out Of meTagenomes (GROOT), and show it to be both faster and more accurate than a current reference-dependent tool for resistome profiling. GROOT runs on a laptop and can process a typical 2 gigabyte metagenome in 2 min using a single CPU. Our method is not restricted to resistome profiling and has the potential to improve current metagenomic workflows.
GROOT is written in Go and is available at https://github.com/will-rowe/groot (MIT license).
Supplementary data are available at Bioinformatics online.
抗菌药物耐药性(AMR)仍然是对全球健康的主要威胁。对宏基因组(“耐药组”)中的集体 AMR 基因进行分析有助于更好地了解 AMR 基因的多样性和动态。反过来,这可以实现对基因的监测、细菌感染的个体化治疗以及更可持续地使用抗菌药物。然而,由于参考基因之间的高度相似性,以及测序数据的巨大数量和分析工作流程的复杂性,耐药组分析可能会变得复杂。我们已经开发出一种有效的、准确的耐药组分析方法,可解决这些复杂问题,并改进现有的工具。
我们的方法将基因集的变体图表示与局部敏感哈希 Forest 索引方案相结合,以允许使用相似性搜索查询快速对宏基因组序列读取进行分类。对分类读取进行后续的层次局部比对和图遍历,使用评分方案可以准确地重建全长基因序列。我们提供了我们的实现方法,即图形化 Resistance Out Of meTagenomes(GROOT),并表明它比当前用于耐药组分析的基于参考的工具更快、更准确。GROOT 可以在笔记本电脑上运行,使用单个 CPU 在 2 分钟内处理典型的 2 千兆字节的宏基因组。我们的方法不仅限于耐药组分析,并且有可能改进当前的宏基因组工作流程。
GROOT 是用 Go 编写的,可以在 https://github.com/will-rowe/groot(MIT 许可证)上找到。
补充数据可在 Bioinformatics 在线获得。