Wakeling Emma, McEntagart Meriel, Bruccoleri Michael, Shaw-Smith Charles, Stals Karen L, Wakeling Matthew, Barnicoat Angela, Beesley Clare, Hanson-Kahn Andrea K, Kukolich Mary, Stevenson David A, Campeau Philippe M, Ellard Sian, Elsea Sarah H, Yang Xiang-Jiao, Caswell Richard C
North East Thames Regional Genetics Service, Great Ormond Street Hospital for Children NHS Foundation Trust, Great Ormond Street, London WC1N 3JH, UK.
Medical Genetics, Floor 0 Jenner Wing, St George's University Hospitals NHS Foundation Trust, Cranmer Terrace, London SW17 0RE, UK.
HGG Adv. 2021 Jan 14;2(1):100015. doi: 10.1016/j.xhgg.2020.100015.
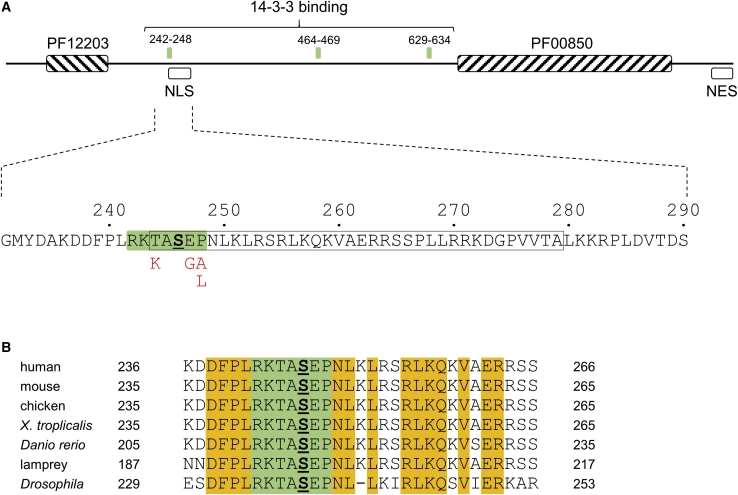
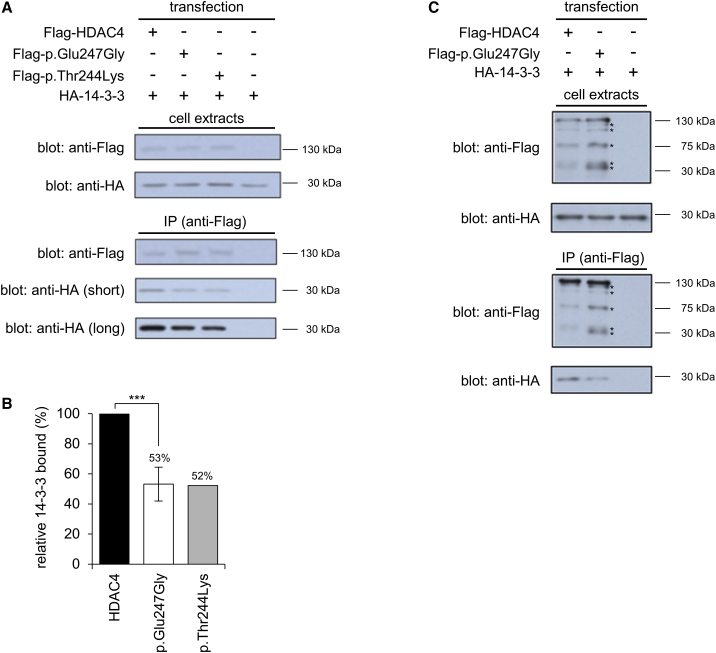
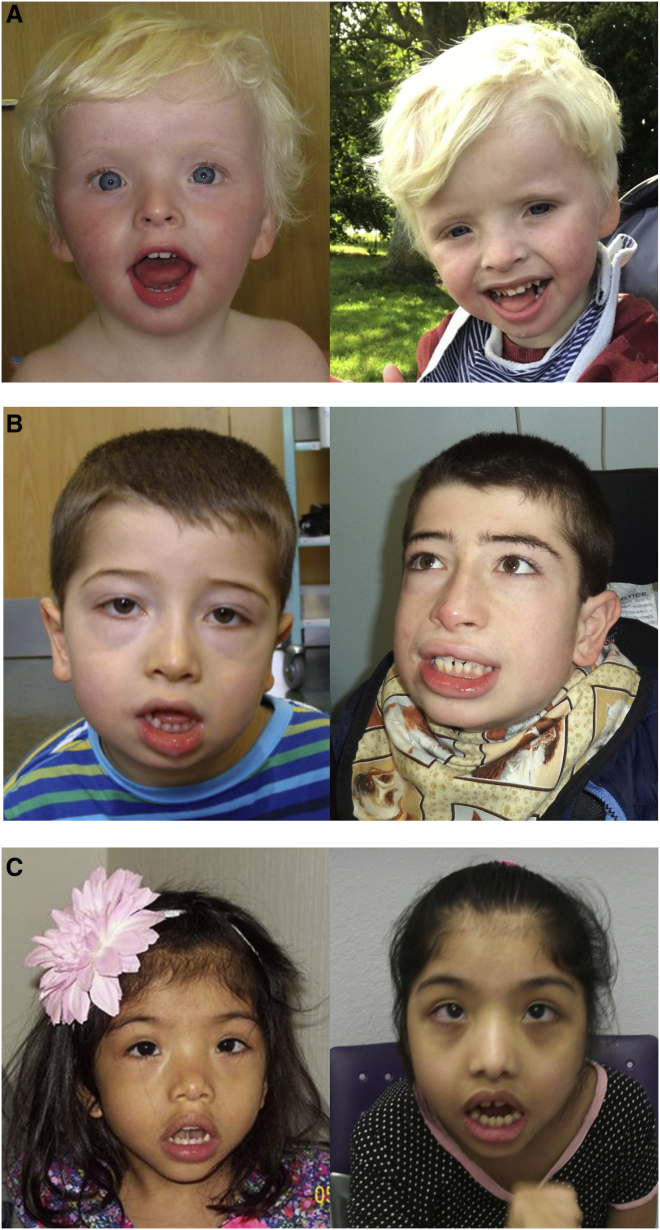
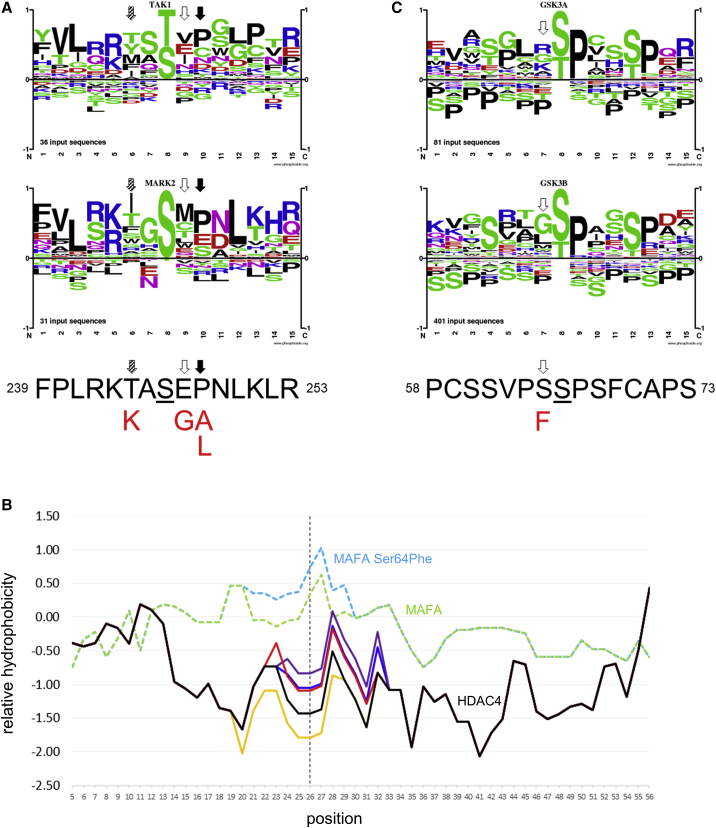
Histone deacetylases play crucial roles in the regulation of chromatin structure and gene expression in the eukaryotic cell, and disruption of their activity causes a wide range of developmental disorders in humans. Loss-of-function alleles of , a founding member of the class IIa deacetylases, have been reported in brachydactyly-mental retardation syndrome (BDMR). However, while disruption of HDAC4 activity and deregulation of its downstream targets may contribute to the BDMR phenotype, loss of HDAC4 function usually occurs as part of larger deletions of chromosome 2q37; BDMR is also known as chromosome 2q37 deletion syndrome, and the precise role of HDAC4 within the phenotype remains uncertain. Thus, identification of missense variants should shed new light on the role of HDAC4 in normal development. Here, we report seven unrelated individuals with a phenotype distinct from that of BDMR, all of whom have heterozygous missense variants that affect a major regulatory site of HDAC4, required for signal-dependent 14-3-3 binding and nucleocytoplasmic shuttling. Two individuals possess variants altering Thr244 or Glu247, whereas the remaining five all carry variants altering Pro248, a key residue for 14-3-3 binding. We propose that the variants in all seven individuals impair 14-3-3 binding (as confirmed for the first two variants by immunoprecipitation assays), thereby identifying deregulation of HDAC4 as a pathological mechanism in a previously uncharacterized developmental disorder.
组蛋白去乙酰化酶在真核细胞的染色质结构和基因表达调控中发挥着关键作用,其活性的破坏会导致人类出现多种发育障碍。IIa类去乙酰化酶的创始成员之一的功能缺失等位基因已在短指-智力发育迟缓综合征(BDMR)中被报道。然而,虽然HDAC4活性的破坏及其下游靶点的失调可能导致BDMR表型,但HDAC4功能的丧失通常是2q37染色体较大缺失的一部分;BDMR也被称为2q37染色体缺失综合征,HDAC4在该表型中的精确作用仍不确定。因此,错义变体的鉴定应为HDAC4在正常发育中的作用提供新的线索。在此,我们报告了7名与BDMR表型不同的无关个体,他们均具有影响HDAC4主要调控位点的杂合错义变体,该位点是信号依赖性14-3-3结合和核质穿梭所必需的。两名个体拥有改变Thr244或Glu247的变体,而其余五名个体均携带改变Pro248的变体,Pro248是14-3-3结合的关键残基。我们提出,所有7名个体中的变体均损害14-3-3结合(前两个变体通过免疫沉淀试验得到证实),从而确定HDAC4失调是一种先前未被描述的发育障碍的病理机制。