Ai Rizi, Boyle David L, Wang Wei, Firestein Gary S
University of California, San Diego.
ACR Open Rheumatol. 2021 Mar;3(3):127-132. doi: 10.1002/acr2.11231. Epub 2021 Feb 5.
To study epigenetic patterns in T lymphocytes that accumulate in the rheumatoid arthritis (RA) synovium, we characterized DNA methylation of CD3 T cells in peripheral blood and synovial tissue in patients with RA and osteoarthritis (OA).
Genomic DNA of CD3 T cells was isolated from patients with RA (n = 8) and OA (n = 5) from blood or the synovium at the time of an arthroplasty using antibodies and magnetic beads. Methylation was measured by using the Illumina Infinium MethylationEPIC Kit. Differentially methylated loci (DML) and differentially methylated genes (DMGs) were identified by using Welch's t-test. Principal component analysis, hierarchical clustering, and pathway analysis were used to determine relationships among groups.
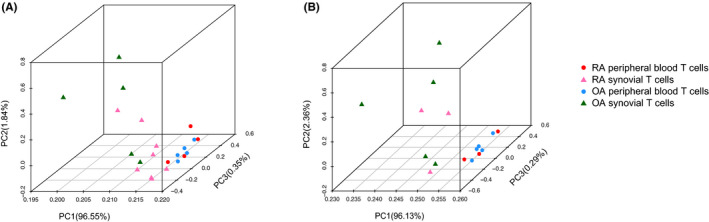
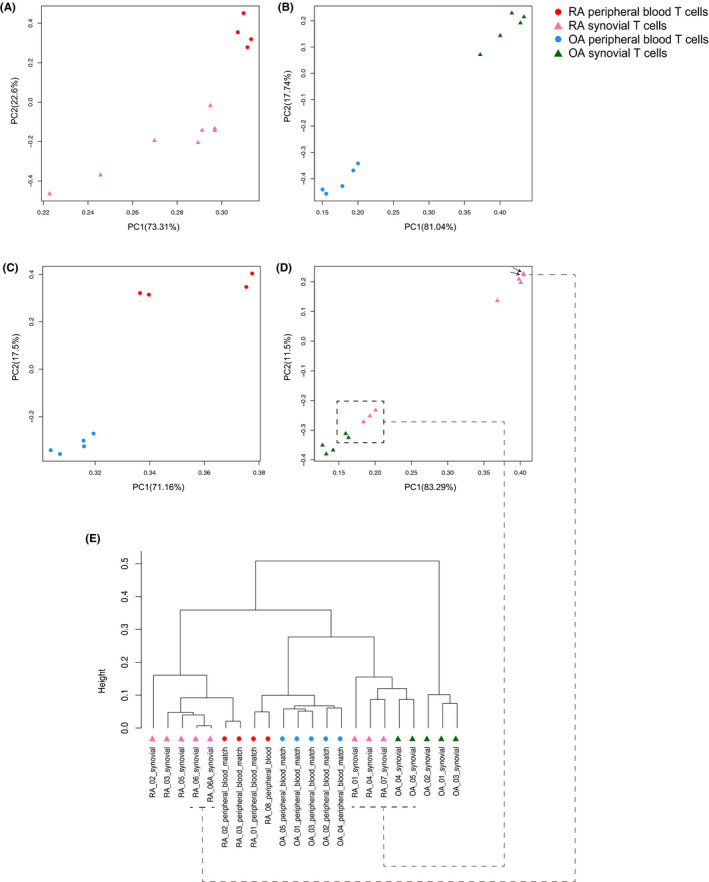
When we compared DNA methylation of CD3 T cells between peripheral blood and synovial tissue within each disease, 4615 and 164 DML were identified in RA and OA samples, respectively, resulting in 832 and 36 DMGs. A principal component analysis showed that methylation differences in T cells were greater on the basis of on location (blood vs synovium) rather than disease (RA vs OA). Differentially modified pathways were significantly enriched between RA blood and synovial T cells, especially in genes related to complement, integrin cell surface interactions, and the P53 pathway. The limited number of DMGs identified between OA blood and synovial T cells did not conform to biologic pathways.
The patterns of DNA methylation in RA show location-specific differences related to immune pathways, whereas methylation differences in OA are limited. The RA joint-specific signatures could be due to selective accumulation of T-cell populations or expansion of differentially marked adaptive immune cells. Understanding epigenetic patterns could provide clues to the types of T cells that accumulate in the RA joint and identify potential therapeutic targets.
为研究在类风湿关节炎(RA)滑膜中积聚的T淋巴细胞的表观遗传模式,我们对RA患者和骨关节炎(OA)患者外周血及滑膜组织中CD3 T细胞的DNA甲基化进行了特征分析。
在关节置换术时,使用抗体和磁珠从RA患者(n = 8)和OA患者(n = 5)的血液或滑膜中分离CD3 T细胞的基因组DNA。使用Illumina Infinium MethylationEPIC试剂盒测量甲基化。通过Welch t检验鉴定差异甲基化位点(DML)和差异甲基化基因(DMG)。使用主成分分析、层次聚类和通路分析来确定各组之间的关系。
当我们比较每种疾病外周血和滑膜组织中CD3 T细胞的DNA甲基化时,在RA和OA样本中分别鉴定出4615个和164个DML,产生832个和36个DMG。主成分分析表明,T细胞中的甲基化差异基于位置(血液与滑膜)而非疾病(RA与OA)更大。RA血液和滑膜T细胞之间差异修饰的通路显著富集,特别是在与补体、整合素细胞表面相互作用和P53通路相关的基因中。在OA血液和滑膜T细胞之间鉴定出的有限数量的DMG不符合生物学通路。
RA中的DNA甲基化模式显示出与免疫通路相关的位置特异性差异,而OA中的甲基化差异有限。RA关节特异性特征可能是由于T细胞群体的选择性积聚或差异标记的适应性免疫细胞的扩增。了解表观遗传模式可为积聚在RA关节中的T细胞类型提供线索,并确定潜在的治疗靶点。