Department of Ophthalmology and Biomedical Genetics, University of Rochester, Rochester, NY, USA.
Department of Ophthalmology, Stein Eye Institute, Department of Neurobiology, David Geffen School of Medicine, Molecular Biology Institute, Brain Research Institute, University of California, Los Angeles, CA, USA.
Commun Biol. 2021 Feb 5;4(1):161. doi: 10.1038/s42003-021-01682-5.
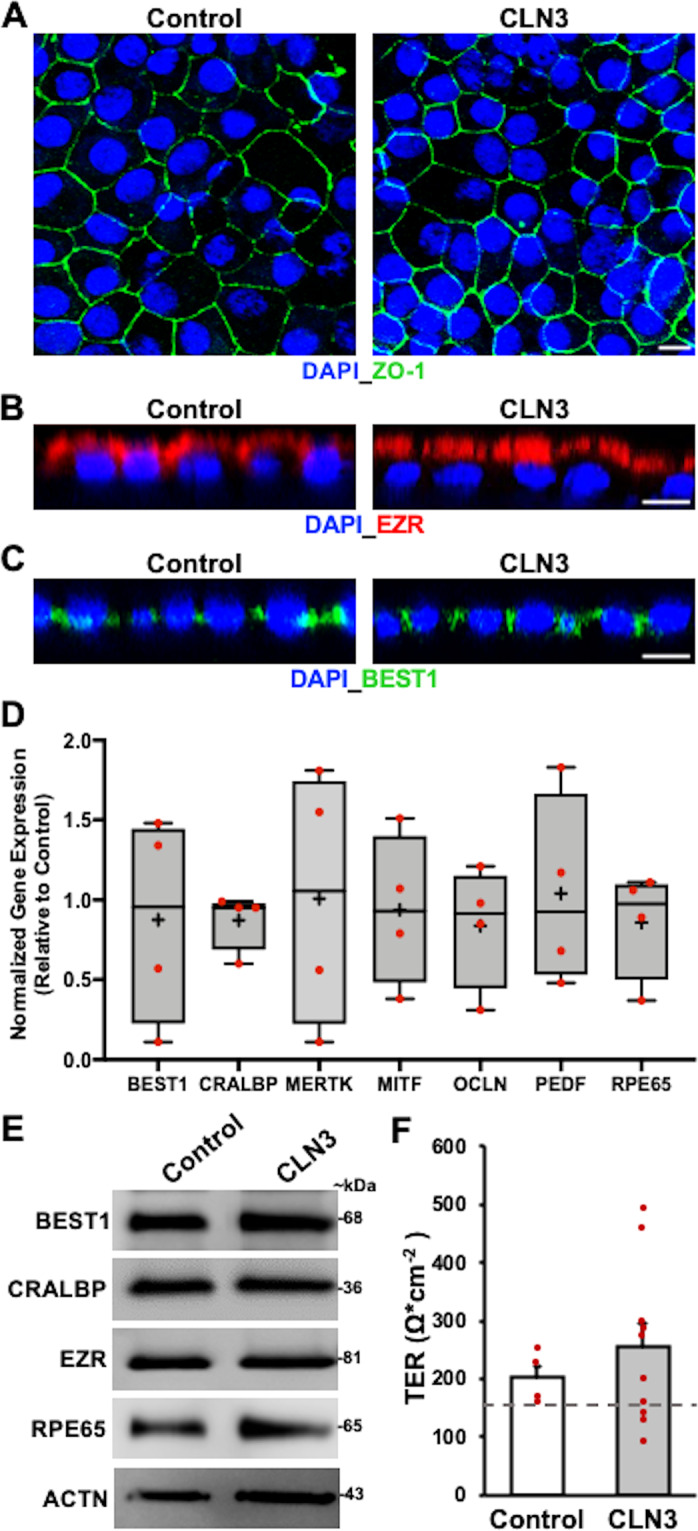
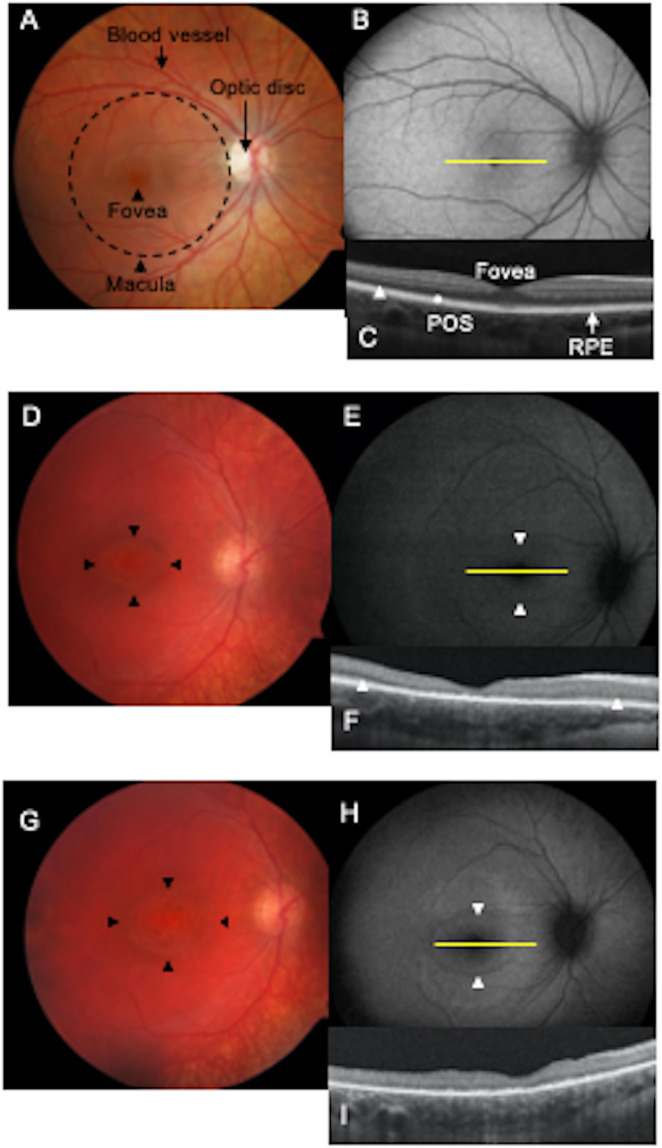
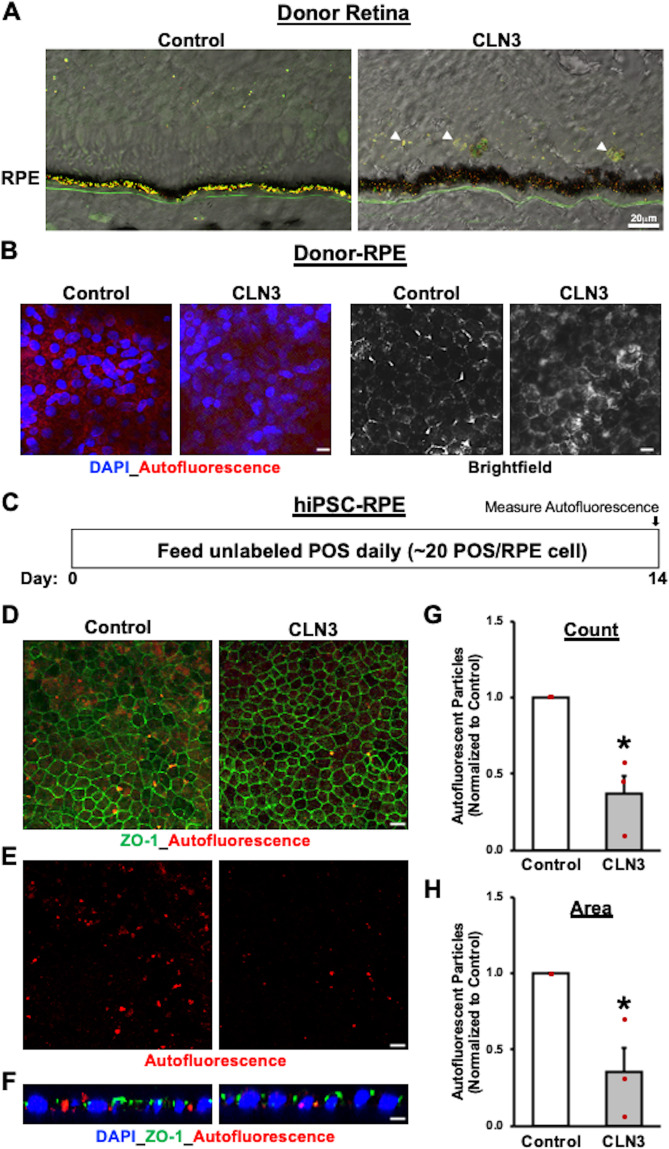
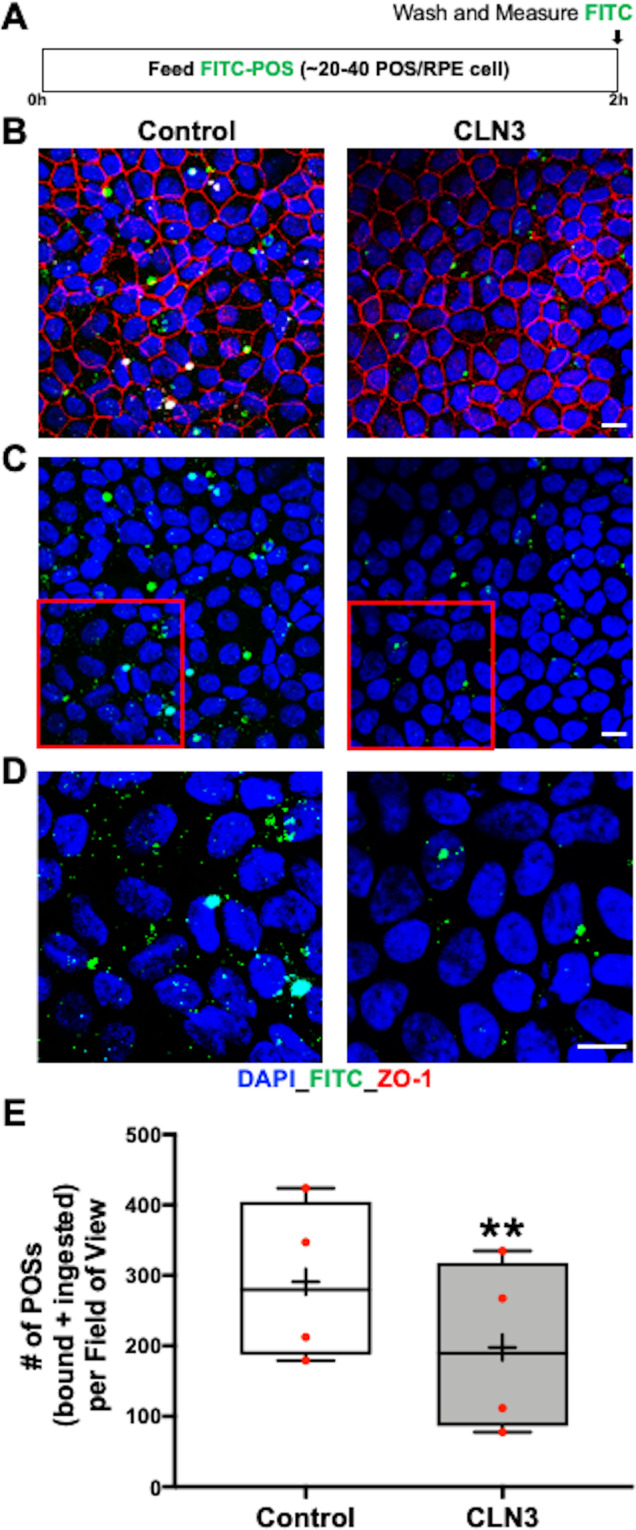
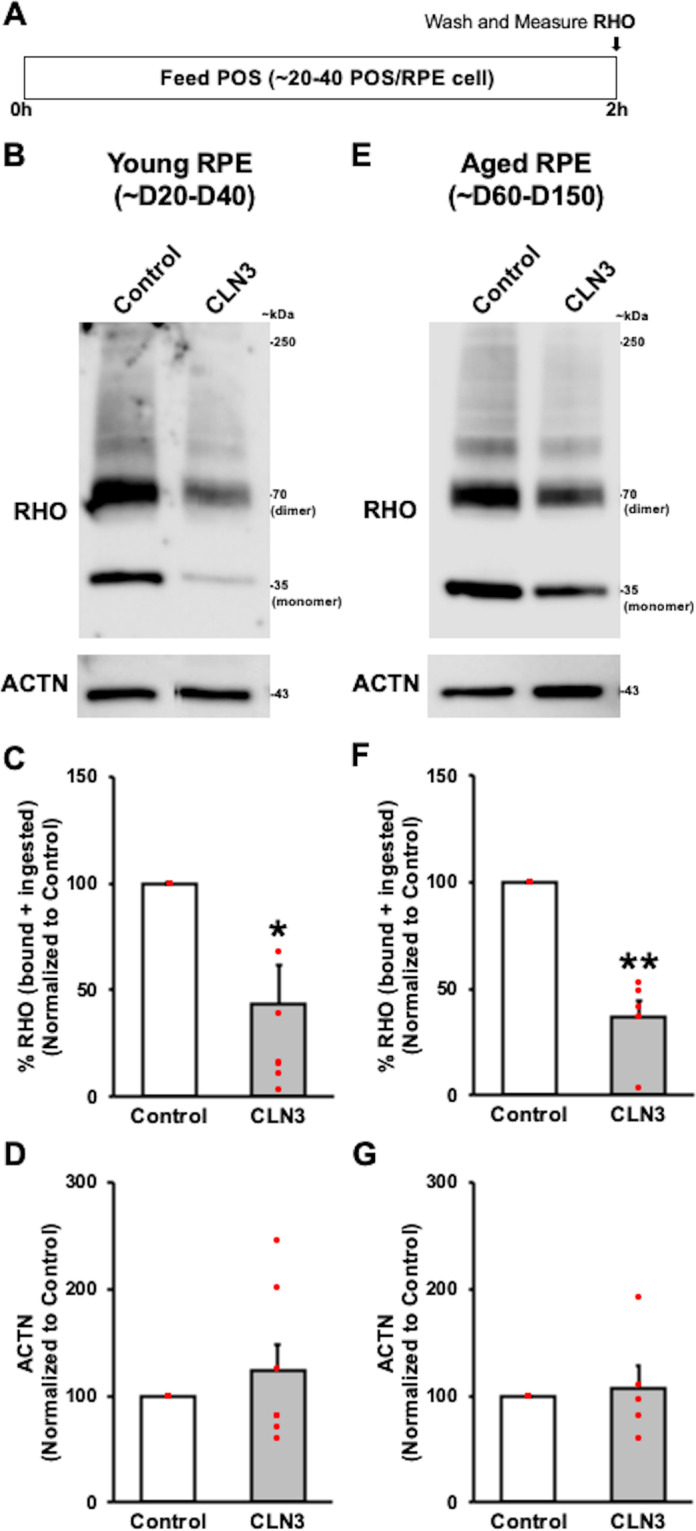
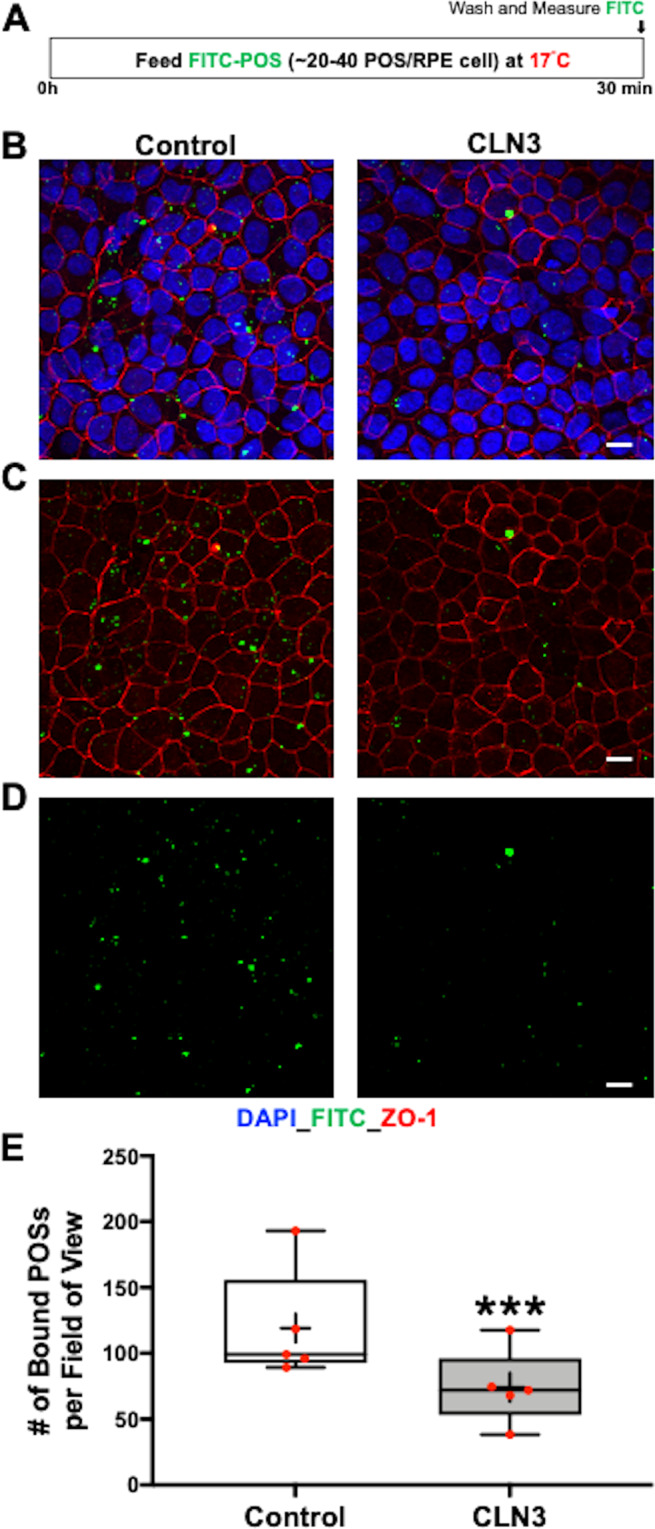
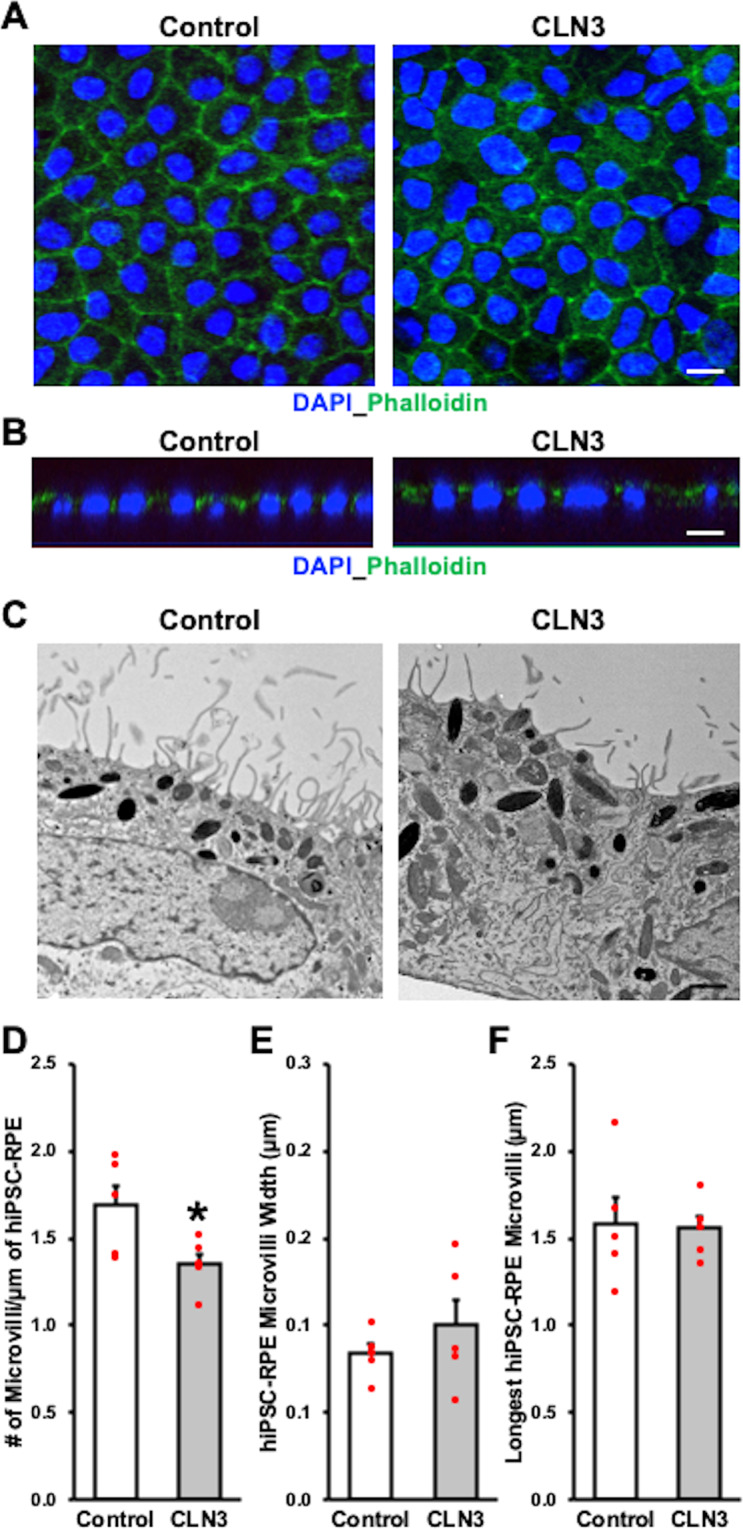
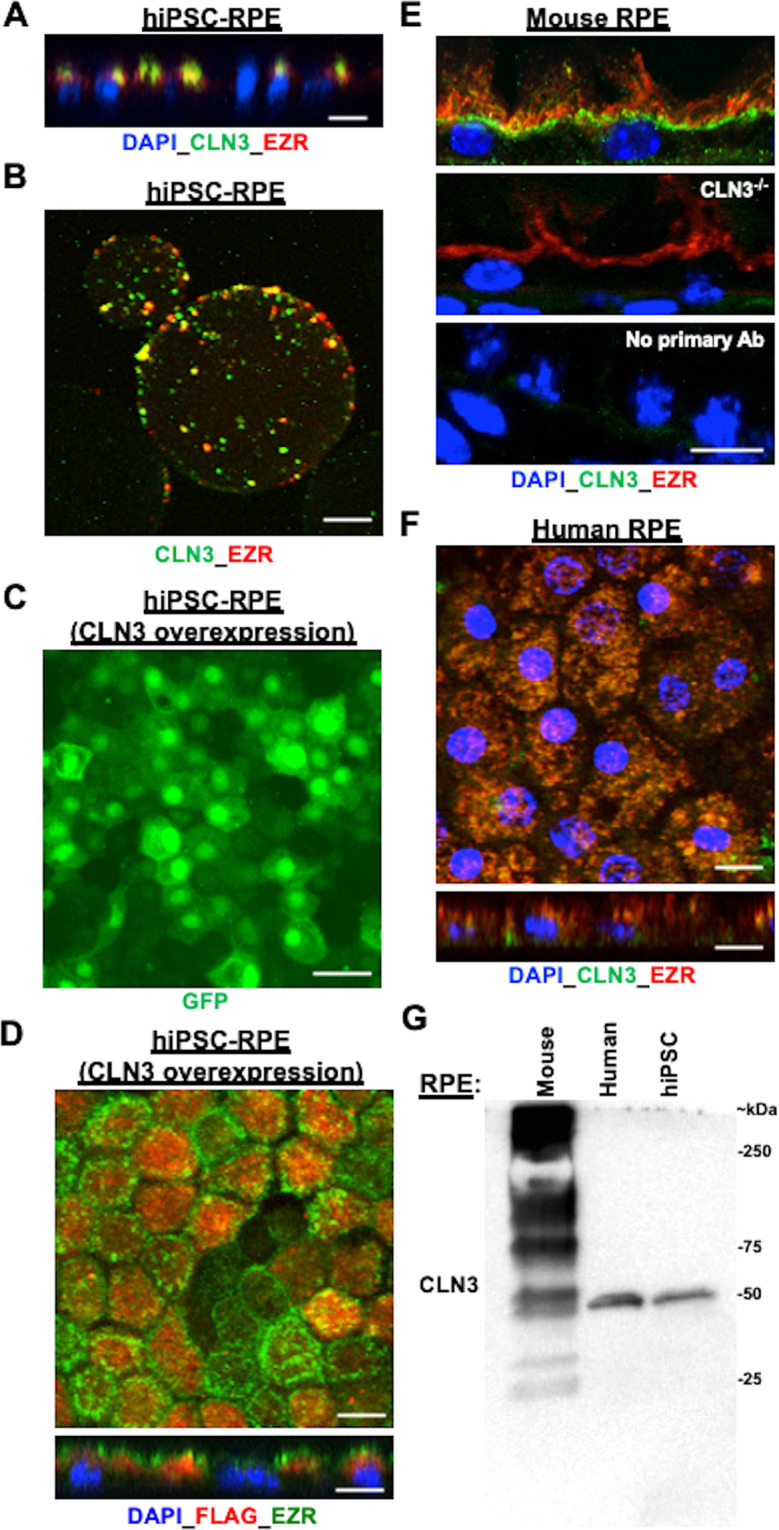
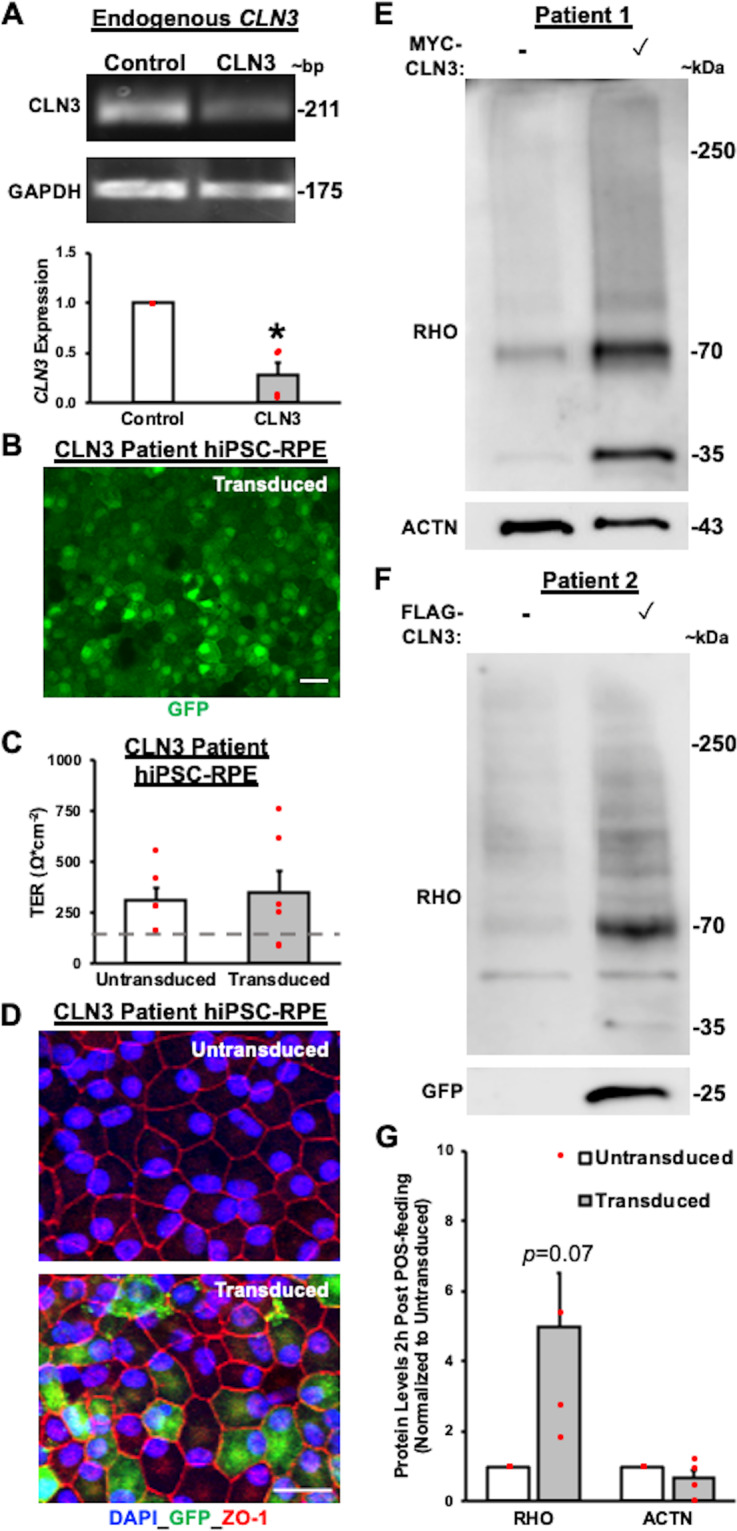
Mutations in CLN3 lead to photoreceptor cell loss in CLN3 disease, a lysosomal storage disorder characterized by childhood-onset vision loss, neurological impairment, and premature death. However, how CLN3 mutations cause photoreceptor cell death is not known. Here, we show that CLN3 is required for phagocytosis of photoreceptor outer segment (POS) by retinal pigment epithelium (RPE) cells, a cellular process essential for photoreceptor survival. Specifically, a proportion of CLN3 in human, mouse, and iPSC-RPE cells localized to RPE microvilli, the site of POS phagocytosis. Furthermore, patient-derived CLN3 disease iPSC-RPE cells showed decreased RPE microvilli density and reduced POS binding and ingestion. Notably, POS phagocytosis defect in CLN3 disease iPSC-RPE cells could be rescued by wild-type CLN3 gene supplementation. Altogether, these results illustrate a novel role of CLN3 in regulating POS phagocytosis and suggest a contribution of primary RPE dysfunction for photoreceptor cell loss in CLN3 disease that can be targeted by gene therapy.
CLN3 基因突变导致 CLN3 病的光感受器细胞丢失,CLN3 病是一种溶酶体贮积症,其特征是儿童期发病的视力丧失、神经功能障碍和过早死亡。然而,CLN3 基因突变如何导致光感受器细胞死亡尚不清楚。在这里,我们表明 CLN3 是视网膜色素上皮(RPE)细胞吞噬光感受器外节(POS)所必需的,这是光感受器存活的关键细胞过程。具体来说,人类、小鼠和 iPSC-RPE 细胞中的一部分 CLN3 定位于 RPE 微绒毛,即 POS 吞噬的部位。此外,来自 CLN3 病患者的 iPSC-RPE 细胞显示出 RPE 微绒毛密度降低以及 POS 结合和吞噬减少。值得注意的是,CLN3 病 iPSC-RPE 细胞中的 POS 吞噬缺陷可以通过野生型 CLN3 基因补充来挽救。总的来说,这些结果说明了 CLN3 在调节 POS 吞噬中的新作用,并表明原发性 RPE 功能障碍可能导致 CLN3 病中的光感受器细胞丢失,这种丢失可以通过基因治疗来靶向。