Hernandez Rolando, Facelli Julio C
Department of Biomedical Informatics and Center for Clinical and Translational Science, The University of Utah, Salt Lake City, Utah, USA.
Heliyon. 2021 Jan 27;7(1):e06013. doi: 10.1016/j.heliyon.2021.e06013. eCollection 2021 Jan.
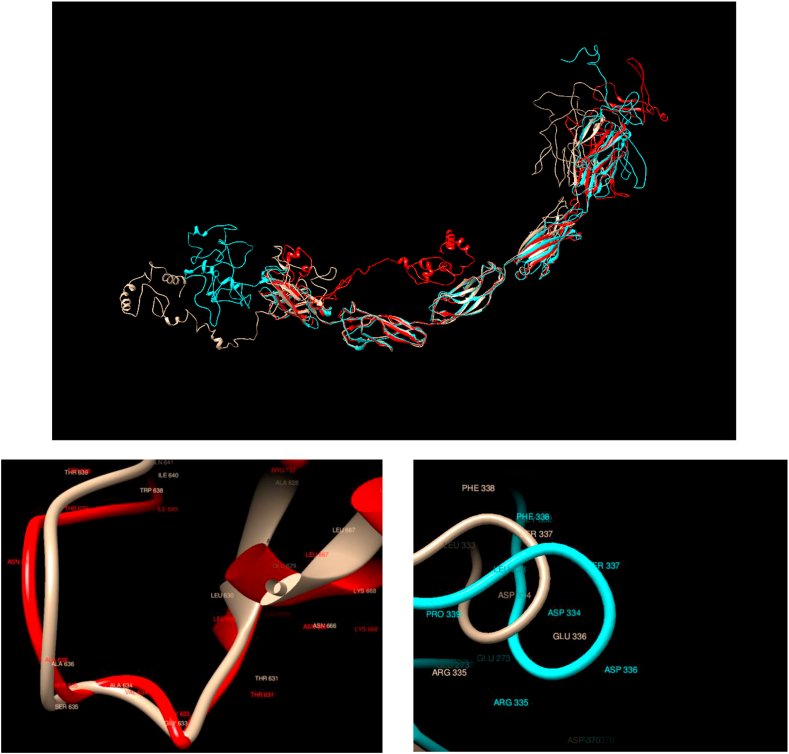
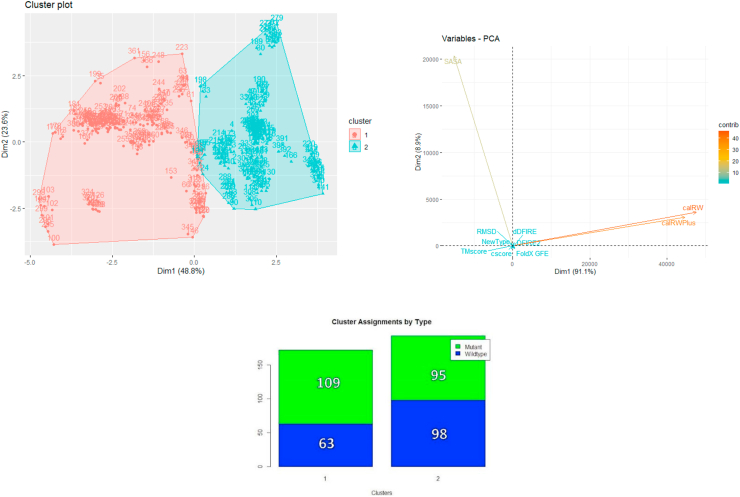
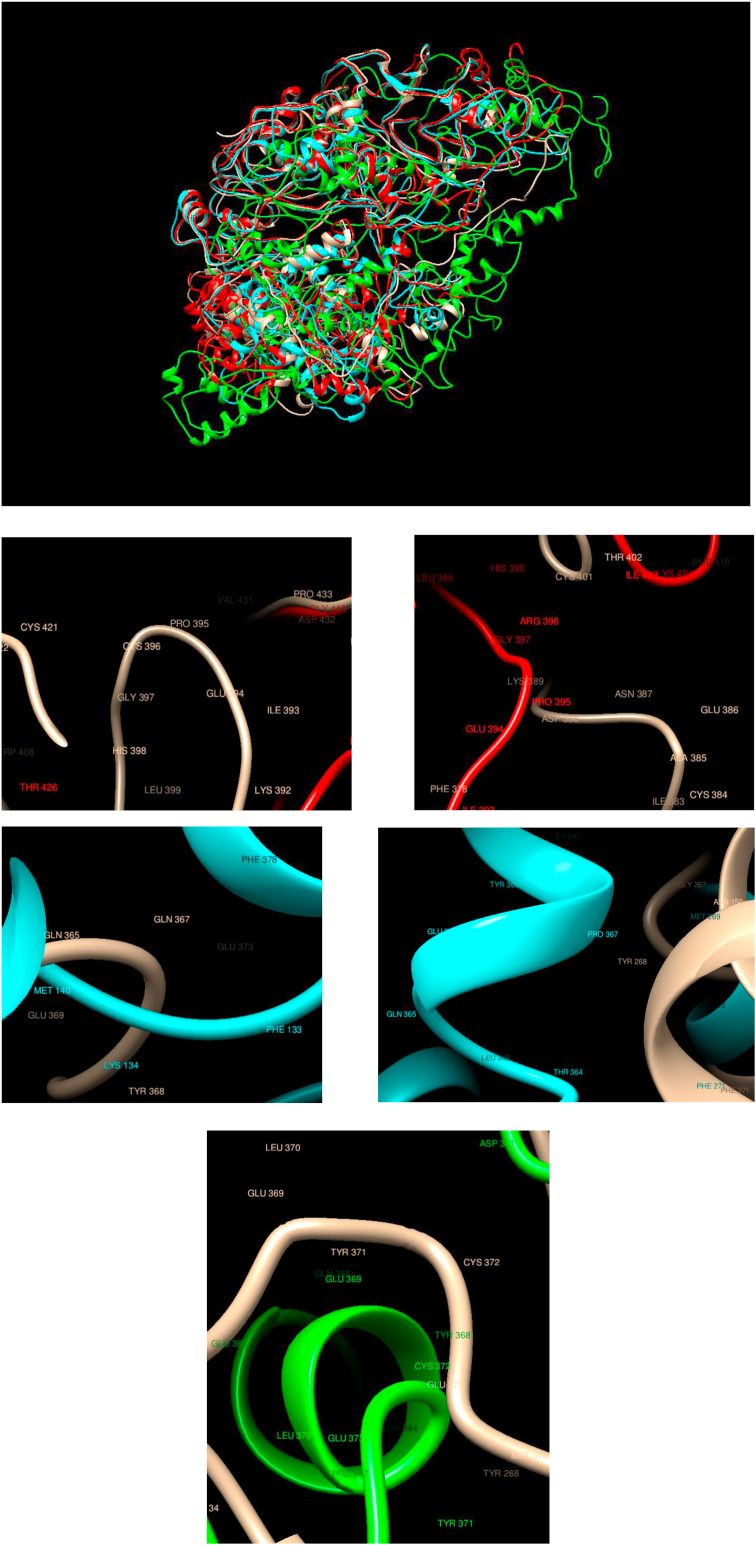
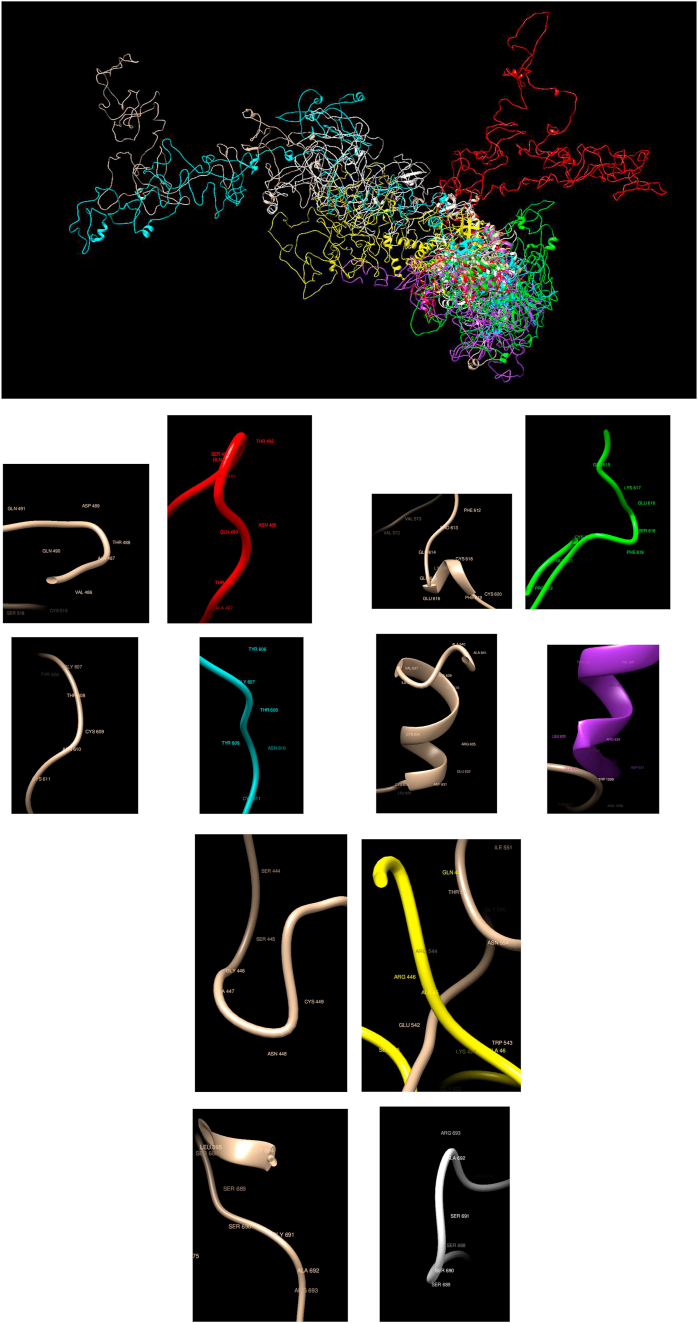
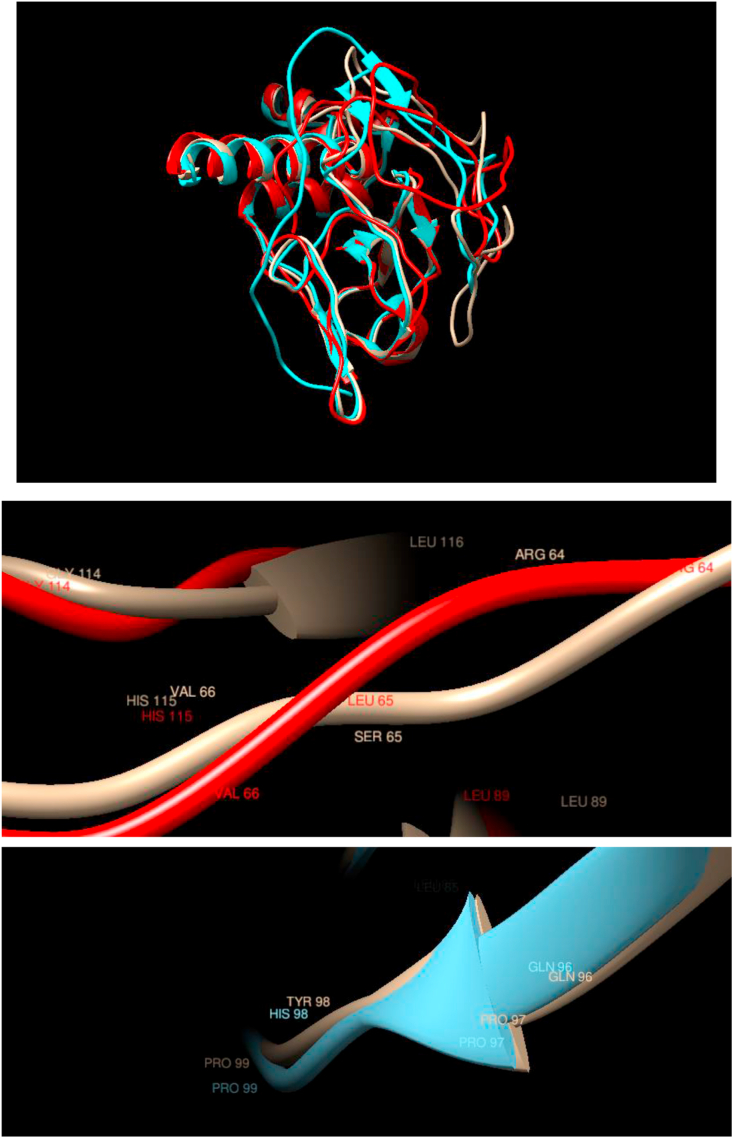
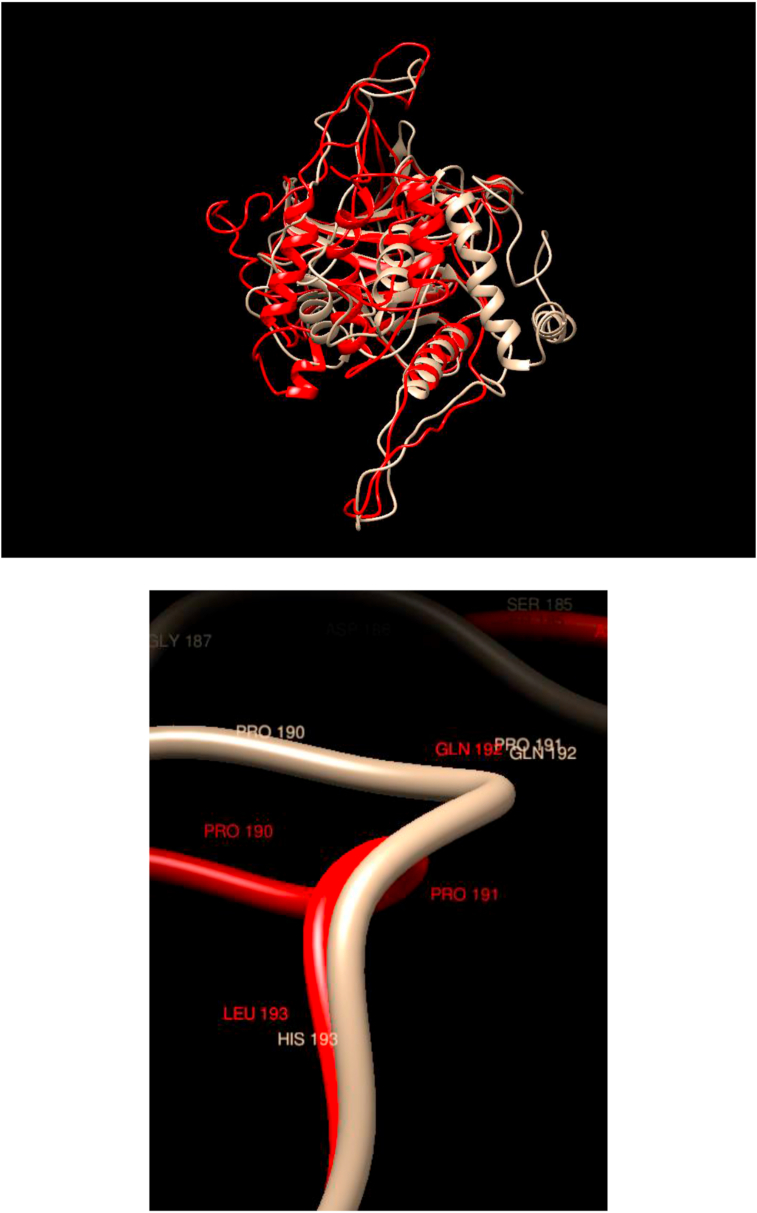
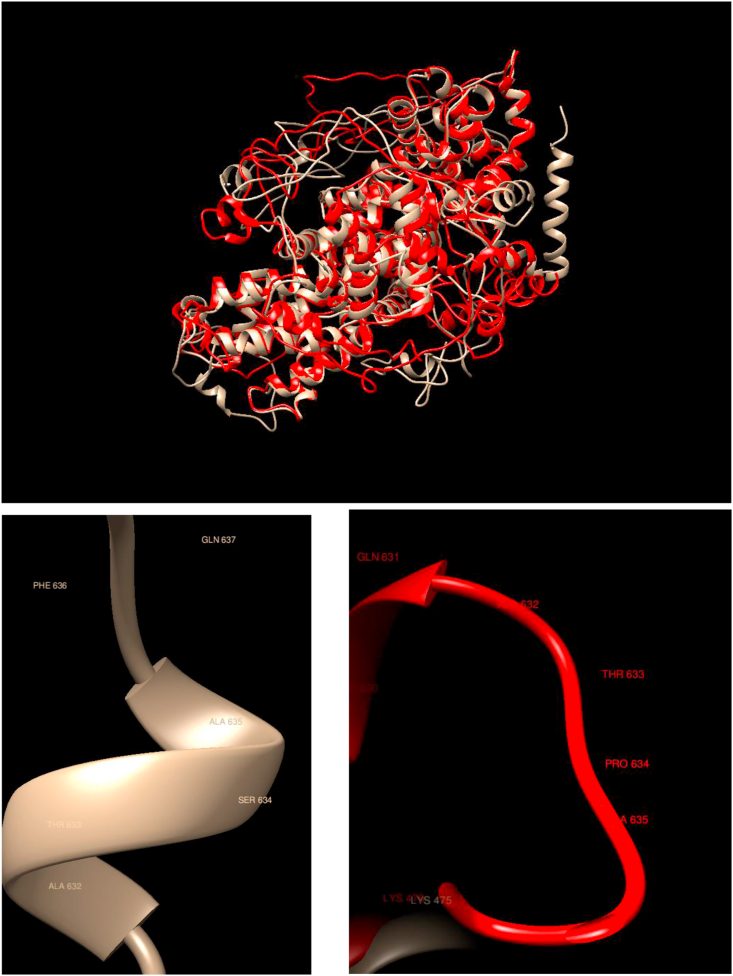
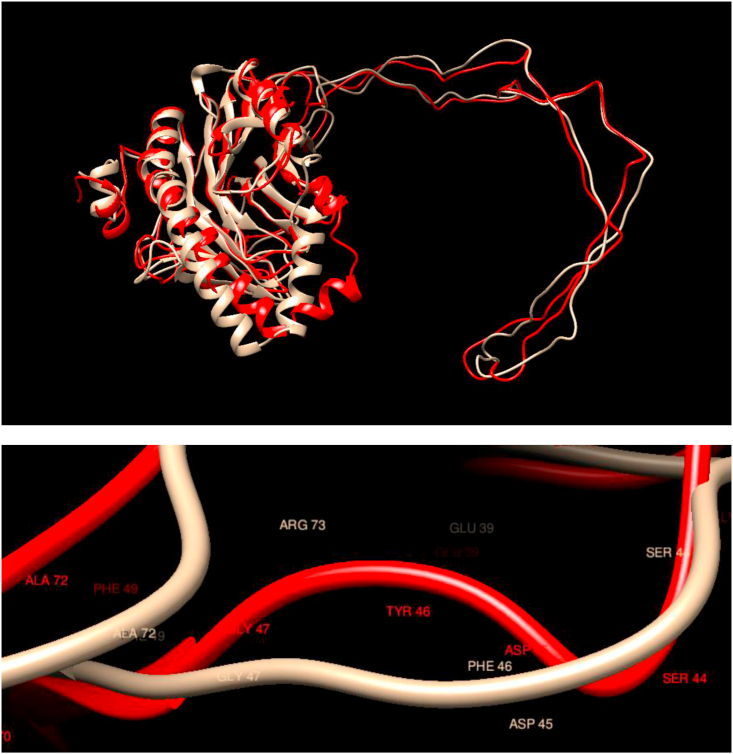
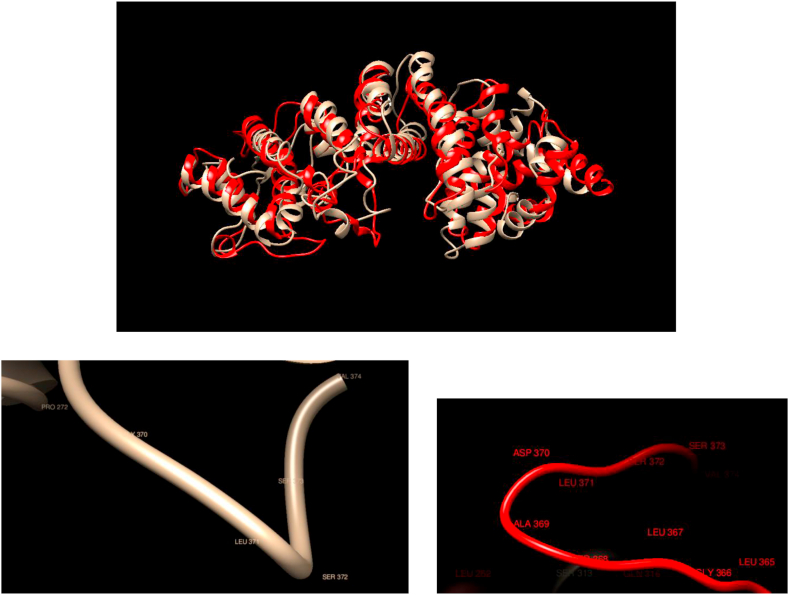
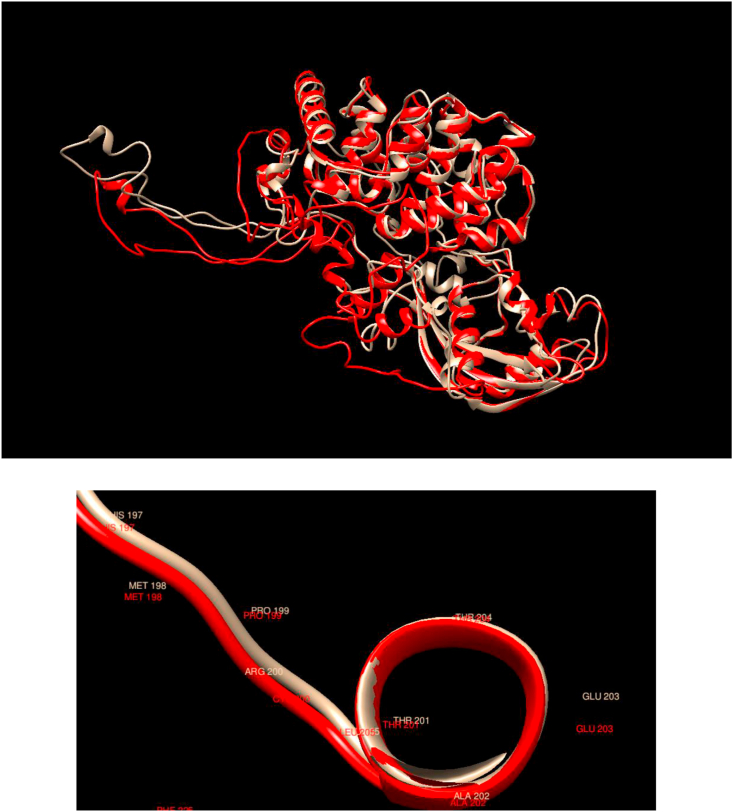
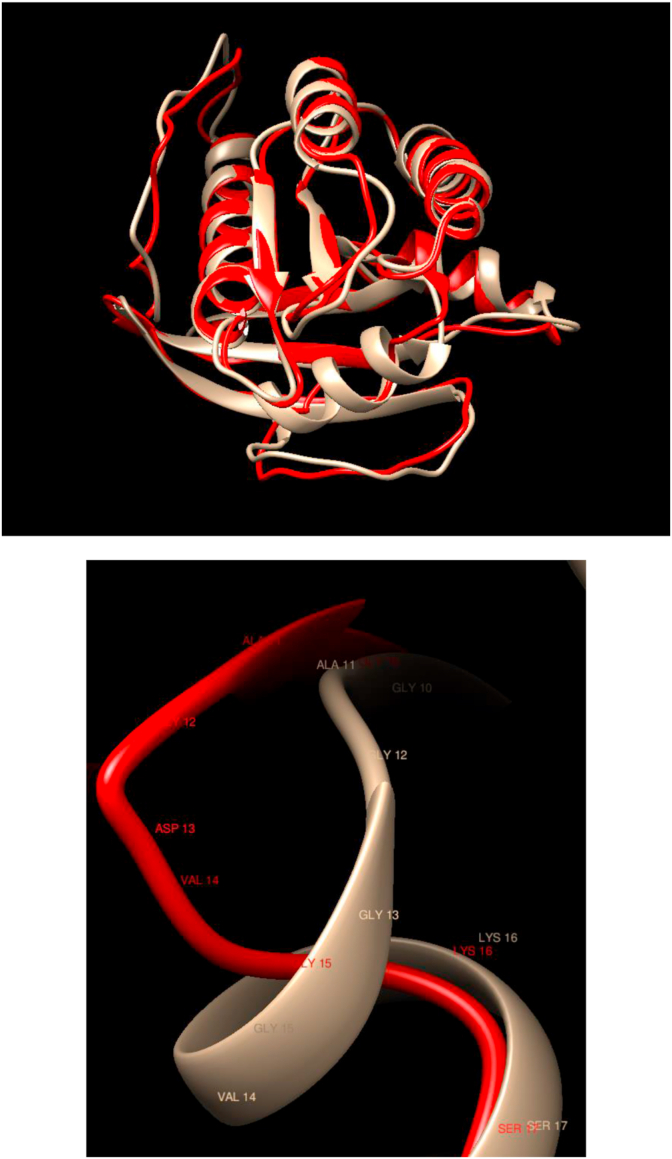
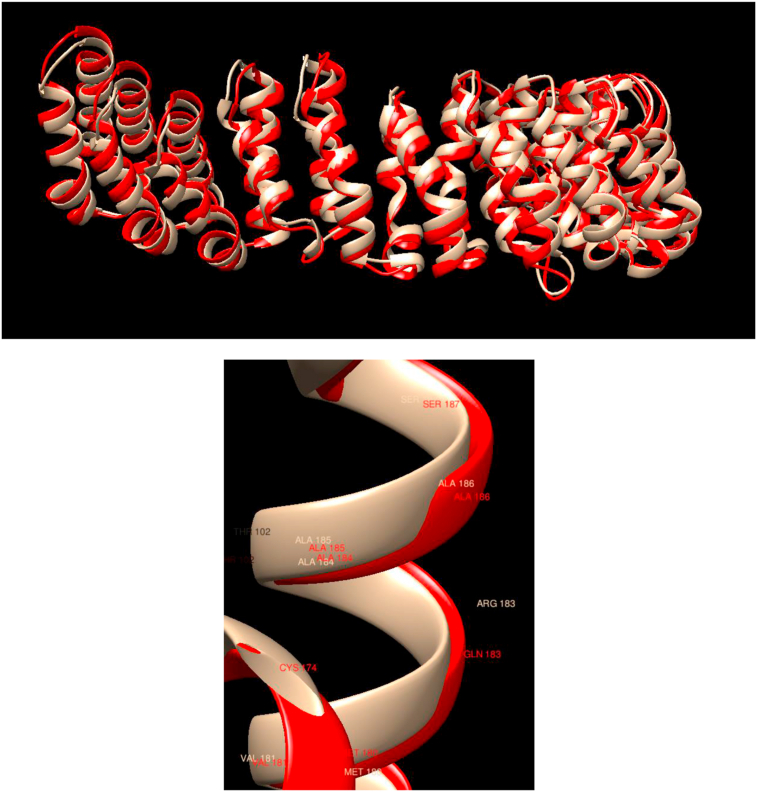
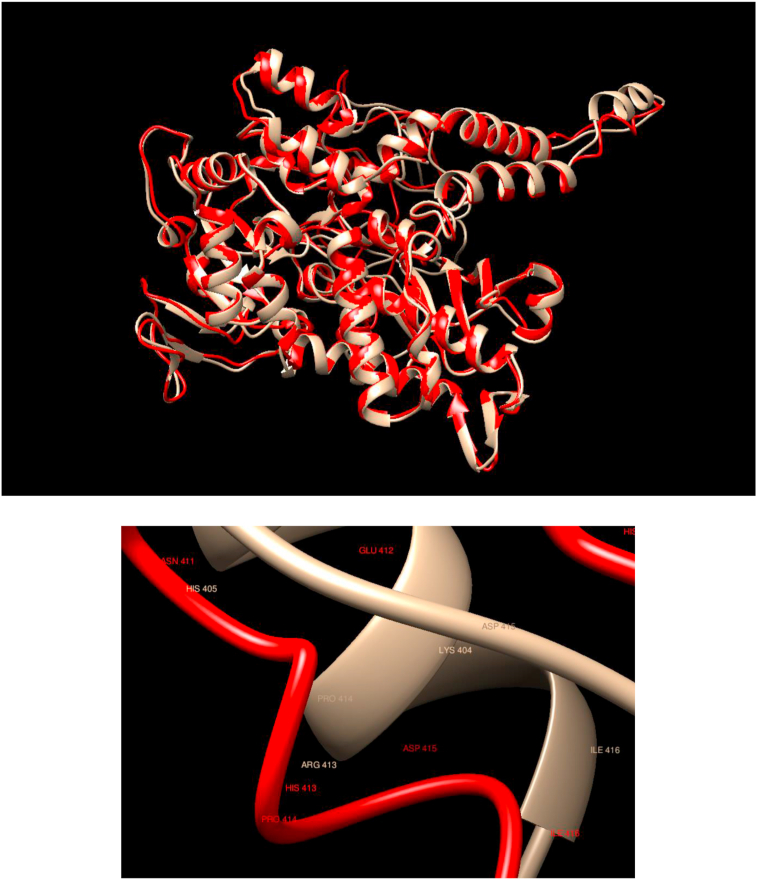
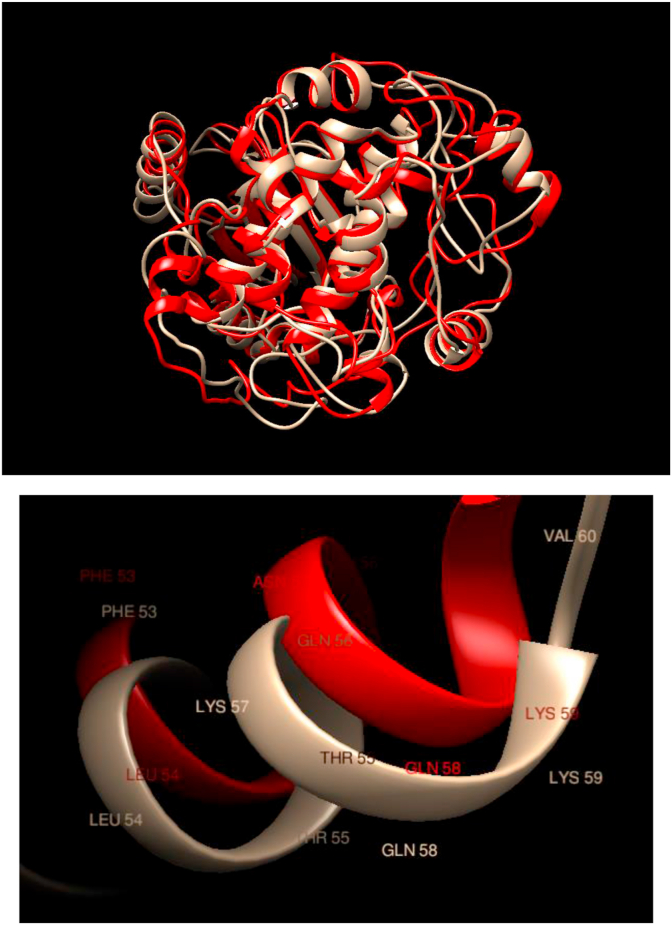
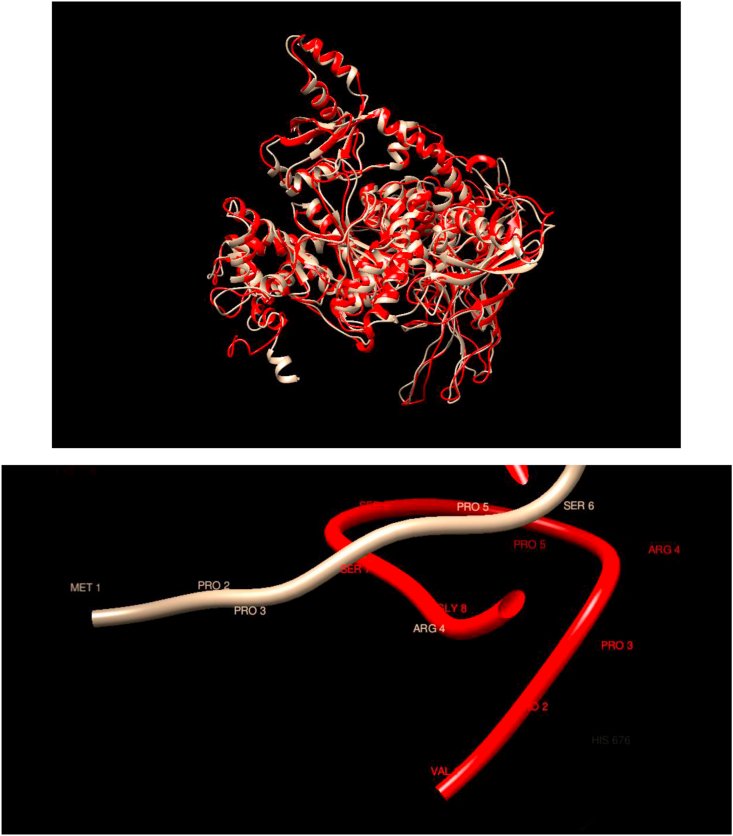
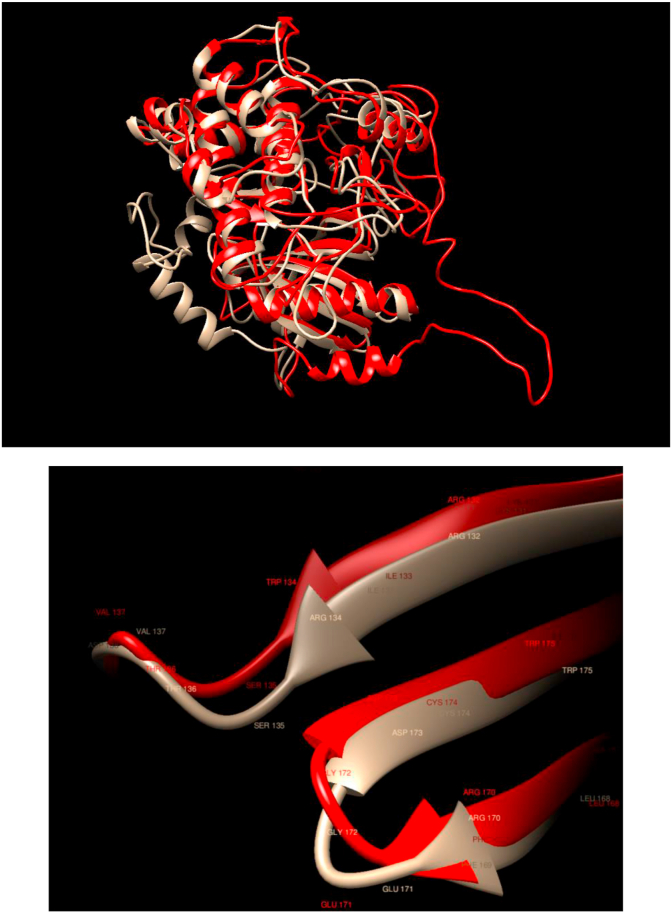
Understanding and predicting the changes of protein structure and function upon mutation and their relationship to human health is a critical element to translate the genomic revolution into actionable interventions. Therefore, it is pertinent to explore how mutations result in structural changes leading to pathogenic proteins, but due to the protein structural knowledge gap, experimental approaches are lacking. Protein structure prediction methods, such as I-TASSER, have made it possible to predict the structure of a given amino acid sequence, thus opening a new way to explore protein structure changes upon mutations when experimental information is not available. Using known mutations from the Catalogue of Somatic Mutation in Cancer (COSMIC) and ClinVar databases, we compare predicted structure-derived properties from wild type (WT) and mutated proteins and find differences between the local and global 3D protein structures of the WT and the mutants. The studies in this relatively small sample reveal that the structural changes are quite diverse.
理解和预测突变后蛋白质结构与功能的变化及其与人类健康的关系,是将基因组革命转化为可行干预措施的关键要素。因此,探索突变如何导致结构变化从而产生致病蛋白至关重要,但由于蛋白质结构知识的空白,缺乏实验方法。诸如I-TASSER之类的蛋白质结构预测方法,使得预测给定氨基酸序列的结构成为可能,从而在缺乏实验信息时开辟了一条探索突变后蛋白质结构变化的新途径。利用来自癌症体细胞突变目录(COSMIC)和临床变异数据库(ClinVar)中的已知突变,我们比较了野生型(WT)和突变蛋白预测的结构衍生特性,并发现了WT和突变体在局部和全局三维蛋白质结构上的差异。在这个相对较小的样本中的研究表明,结构变化非常多样。