de la Cuesta-Zuluaga Jacobo, Spector Tim D, Youngblut Nicholas D, Ley Ruth E
Department of Microbiome Science, Max Planck Institute for Developmental Biology, Tübingen, Germany.
Department of Twin Research and Genetic Epidemiology, King's College London, London, UK.
mSystems. 2021 Feb 9;6(1):e00939-20. doi: 10.1128/mSystems.00939-20.
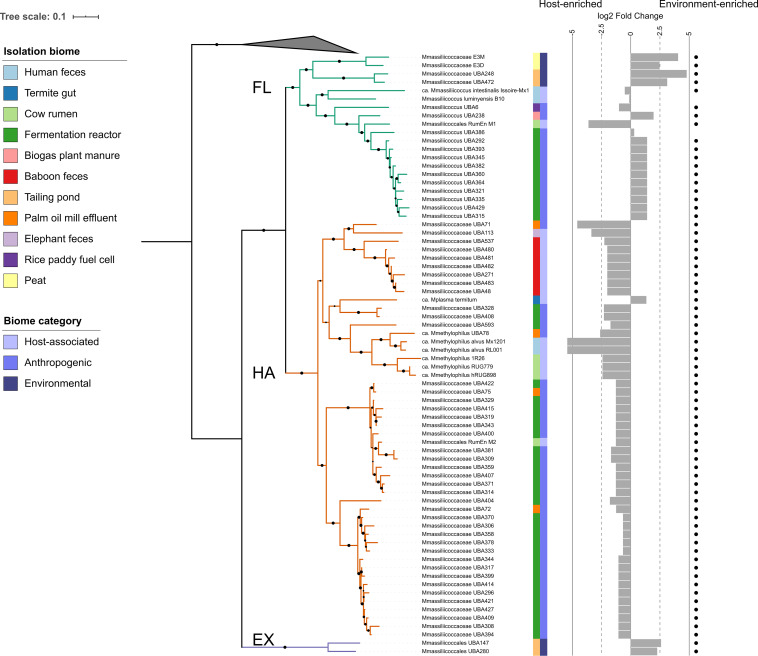
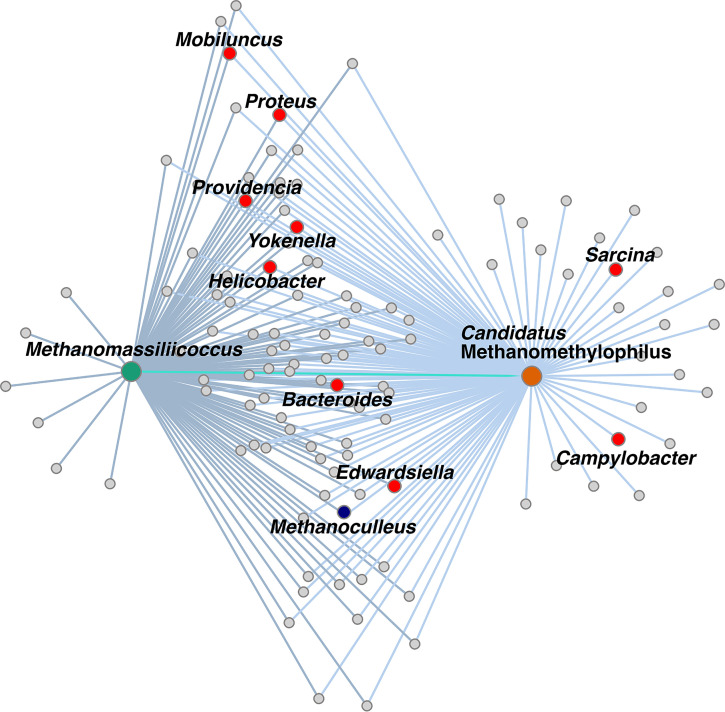
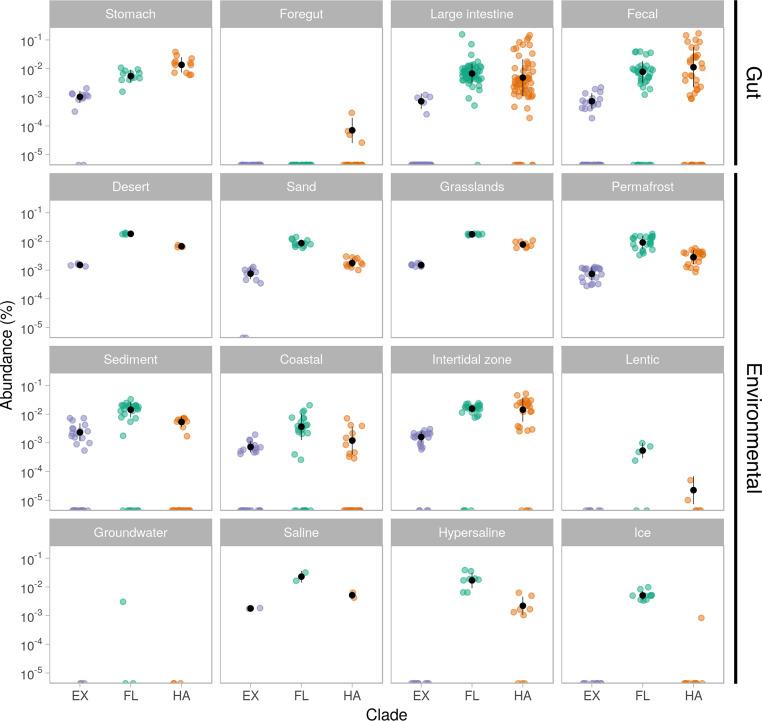
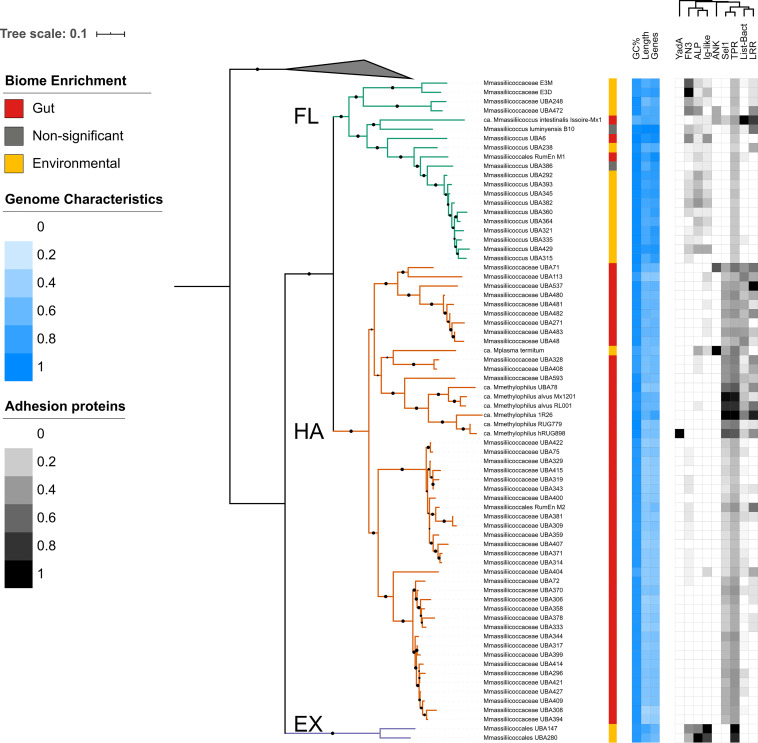
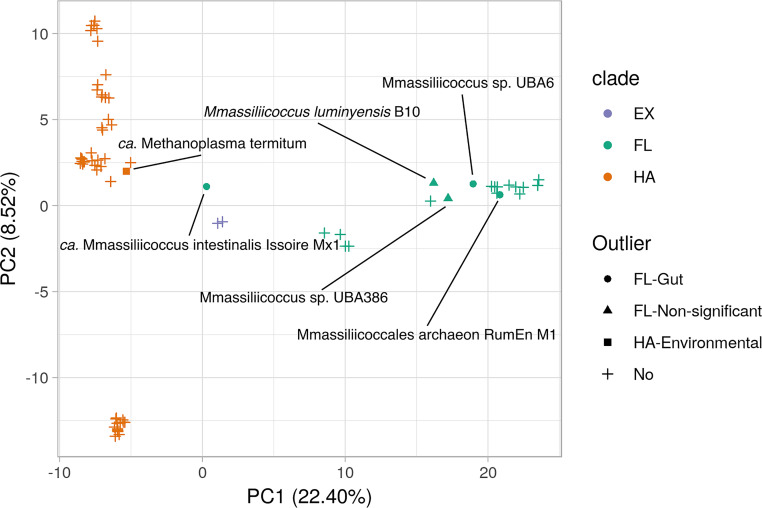
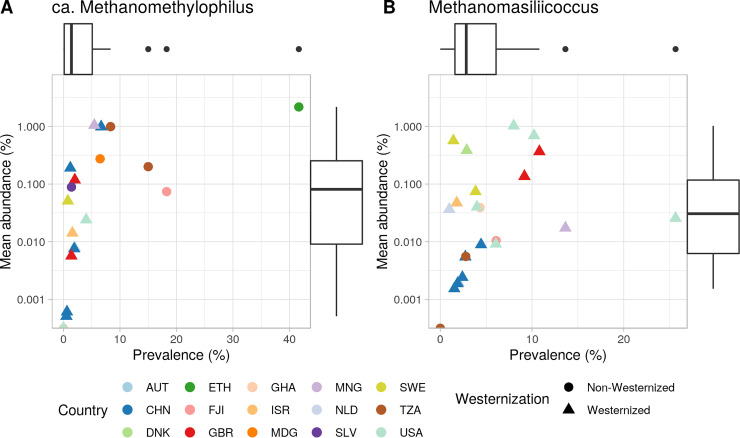
of the order use methylated amines such as trimethylamine as the substrates for methanogenesis. They form two large phylogenetic clades and reside in diverse environments, from soil to the human gut. Two genera, one from each clade, inhabit the human gut: , which has one cultured representative, and " Methanomethylophilus," which has none. Questions remain regarding their distribution across biomes and human populations, their association with other taxa in the gut, and whether host genetics correlate with their abundance. To gain insight into the clade, particularly its human-associated members, we performed a genomic comparison of 72 genomes and assessed their presence in metagenomes derived from the human gut ( = 4,472, representing 22 populations), nonhuman animal gut ( = 145), and nonhost environments ( = 160). Our analyses showed that all taxa are generalists; they were detected in animal gut and environmental samples. We confirmed two large clades, one enriched in the gut and the other enriched in the environment, with notable exceptions. Genomic adaptations to the gut include genome reduction and genes involved in the shikimate pathway and bile resistance. Genomic adaptations differed by clade, not habitat preference, indicating convergent evolution between the clades. In the human gut, the relative abundance of spp. correlated with trimethylamine-producing bacteria and was unrelated to host genotype. Our results shed light on the microbial ecology of this group and may help guide -based strategies for trimethylamine mitigation in cardiovascular disease. are less-known members of the human gut archaeome. Members of this order use methylated amines, including trimethylamine, in methane production. This group has only one cultured representative; how its members adapted to inhabit the mammalian gut and how they interact with other microbes is largely unknown. Using bioinformatics methods applied to DNA from a wide range of samples, we profiled the abundances of these spp. in environmental and host-associated microbial communities. We observed two groups of , one largely host associated and one largely found in environmental samples, with some exceptions. When host associated, these have smaller genomes and possess genes related to bile resistance and aromatic amino acid precursors. We did not detect in all human populations tested, but when present, they were correlated with bacteria known to produce trimethylamine. Due to their metabolism of trimethylamine, these intriguing may form the basis of novel therapies for cardiovascular disease.
该目利用甲基化胺类(如三甲胺)作为产甲烷的底物。它们形成两个大型系统发育分支,存在于从土壤到人类肠道等各种不同环境中。有两个属,每个分支各有一个,栖息于人类肠道:一个属有一个已培养的代表菌种,另一个属“嗜甲基ophilus甲烷菌属”则没有。关于它们在不同生物群落和人类群体中的分布、它们与肠道中其他分类群的关联,以及宿主遗传学是否与它们的丰度相关,仍然存在问题。为了深入了解这个分支,特别是其与人类相关的成员,我们对72个该目基因组进行了比较,并评估了它们在源自人类肠道(n = 4472,代表22个群体)、非人类动物肠道(n = 145)和非宿主环境(n = 160)的宏基因组中的存在情况。我们的分析表明,所有分类群都是广适性的;它们在动物肠道和环境样本中均被检测到。我们确认了两个大型分支,一个在肠道中富集,另一个在环境中富集,但有显著例外。对肠道的基因组适应性包括基因组缩减以及与莽草酸途径和胆汁抗性相关的基因。基因组适应性因分支而异,而非因栖息地偏好而异,这表明分支之间存在趋同进化。在人类肠道中,该目某些菌种的相对丰度与产生三甲胺的细菌相关,且与宿主基因型无关。我们的结果揭示了这一群体的微生物生态学,可能有助于指导基于该目的策略来减轻心血管疾病中的三甲胺水平。该目是人类肠道古菌群落中鲜为人知的成员。该目成员在甲烷生成过程中利用甲基化胺类,包括三甲胺。这个群体只有一个已培养的代表菌种;其成员如何适应栖息于哺乳动物肠道以及它们如何与其他微生物相互作用在很大程度上尚不清楚。我们使用应用于来自广泛样本的DNA的生物信息学方法,分析了这些该目菌种在环境和宿主相关微生物群落中的丰度。我们观察到两组该目菌种,一组主要与宿主相关,另一组主要存在于环境样本中,但有一些例外情况。当与宿主相关时,这些该目菌种的基因组较小,并拥有与胆汁抗性和芳香族氨基酸前体相关的基因。在我们测试的所有人类群体中并非都检测到该目菌种,但当存在时,它们与已知产生三甲胺的细菌相关。由于它们对三甲胺的代谢,这些有趣的该目菌种可能构成心血管疾病新疗法的基础。