de Oliveira Martins Leonardo, Page Andrew J, Mather Alison E, Charles Ian G
Quadram Institute Bioscience, Norwich Research Park, Norwich, NR4 7UQ, UK.
Faculty of Medicine and Health Sciences, University of East Anglia, Norwich, NR4 7TJ, UK.
NAR Genom Bioinform. 2019 Nov 14;2(1):lqz016. doi: 10.1093/nargab/lqz016. eCollection 2020 Mar.
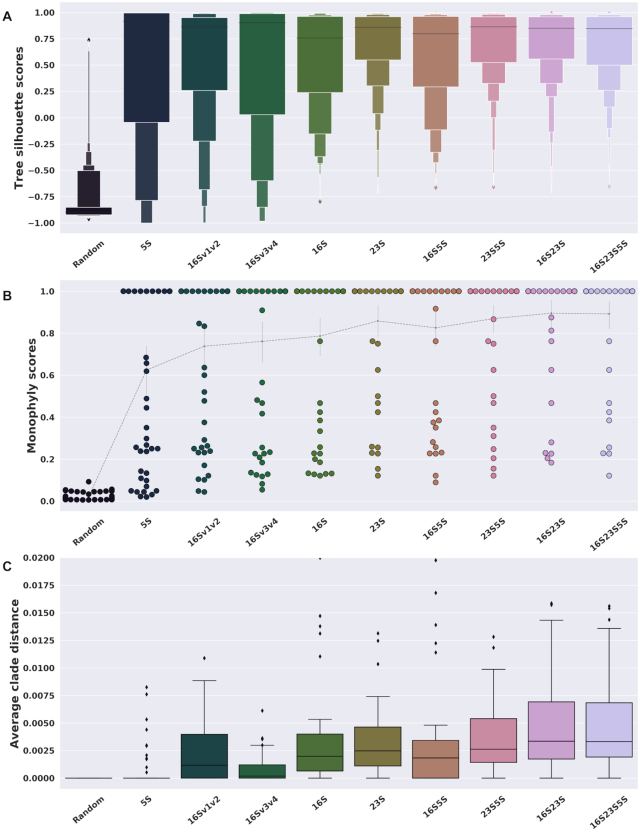
DNA barcoding through the use of amplified regions of the ribosomal operon, such as the 16S gene, is a routine method to gain an overview of the microbial taxonomic diversity within a sample without the need to isolate and culture the microbes present. However, bacterial cells usually have multiple copies of this ribosomal operon, and choosing the 'wrong' copy could provide a misleading species classification. While this presents less of a problem for well-characterized organisms with large sequence databases to interrogate, it is a significant challenge for lesser known organisms with unknown copy number and diversity. Using the entire length of the ribosomal operon, which encompasses the 16S, 23S, 5S and internal transcribed spacer regions, should provide greater taxonomic resolution but has not been well explored. Here, we use publicly available reference genomes and explore the theoretical boundaries when using concatenated genes and the full-length ribosomal operons, which has been made possible by the development and uptake of long-read sequencing technologies. We quantify the issues of both copy choice and operon length in a phylogenetic context to demonstrate that longer regions improve the phylogenetic signal while maintaining taxonomic accuracy.
通过使用核糖体操纵子的扩增区域(如16S基因)进行DNA条形码分析,是一种无需分离和培养样本中存在的微生物就能全面了解微生物分类多样性的常规方法。然而,细菌细胞通常有多个核糖体操纵子拷贝,选择“错误”的拷贝可能会导致误导性的物种分类。虽然对于有大量序列数据库可供查询的特征明确的生物体来说,这不是什么大问题,但对于拷贝数和多样性未知的鲜为人知的生物体而言,这是一个重大挑战。使用核糖体操纵子的全长,包括16S、23S、5S和内部转录间隔区,应该能提供更高的分类分辨率,但尚未得到充分探索。在这里,我们利用公开可用的参考基因组,探索使用串联基因和全长核糖体操纵子时的理论界限,长读长测序技术的发展和应用使这成为可能。我们在系统发育背景下量化了拷贝选择和操纵子长度的问题,以证明更长的区域在保持分类准确性的同时提高了系统发育信号。