Dohm Juliane C, Peters Philipp, Stralis-Pavese Nancy, Himmelbauer Heinz
Institute of Computational Biology, Department of Biotechnology, University of Life Sciences and Natural Resources, Vienna (BOKU), Muthgasse 18, 1190 Vienna, Austria.
NAR Genom Bioinform. 2020 May 25;2(2):lqaa037. doi: 10.1093/nargab/lqaa037. eCollection 2020 Jun.
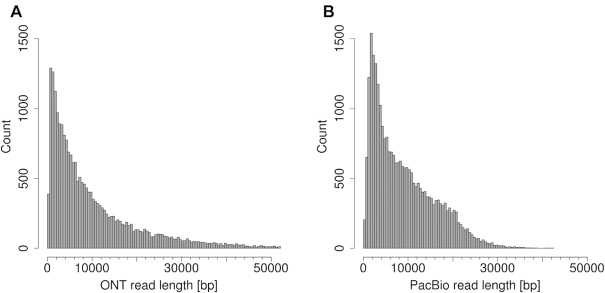
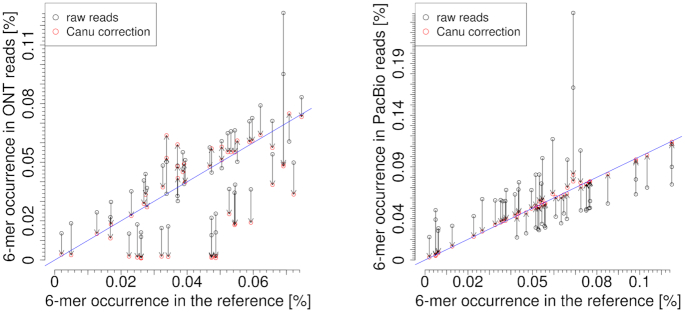
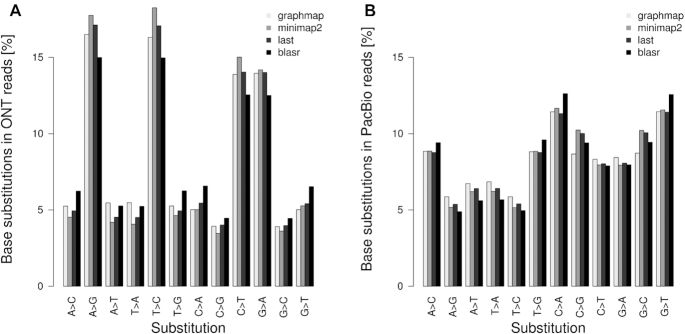
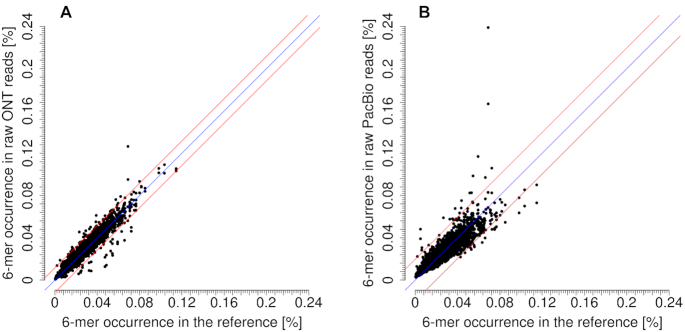
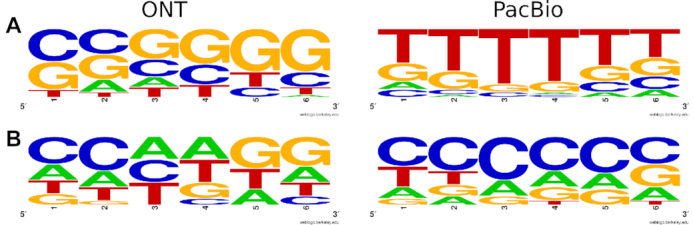
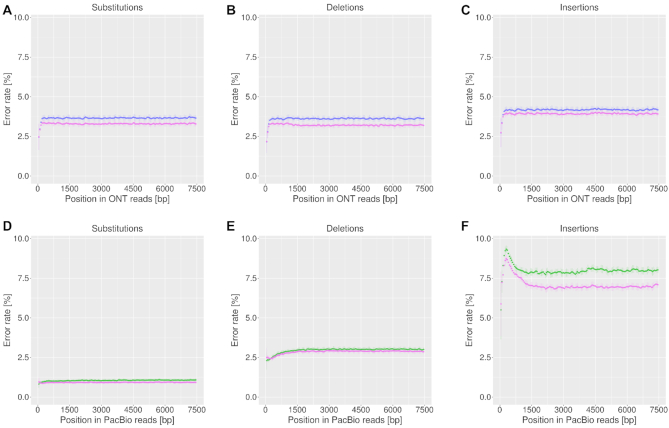
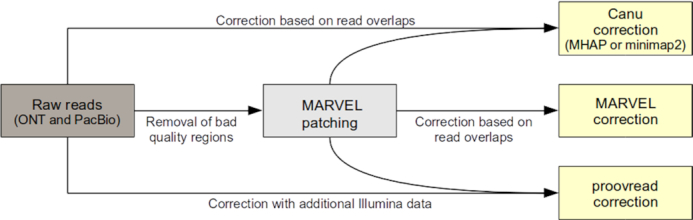
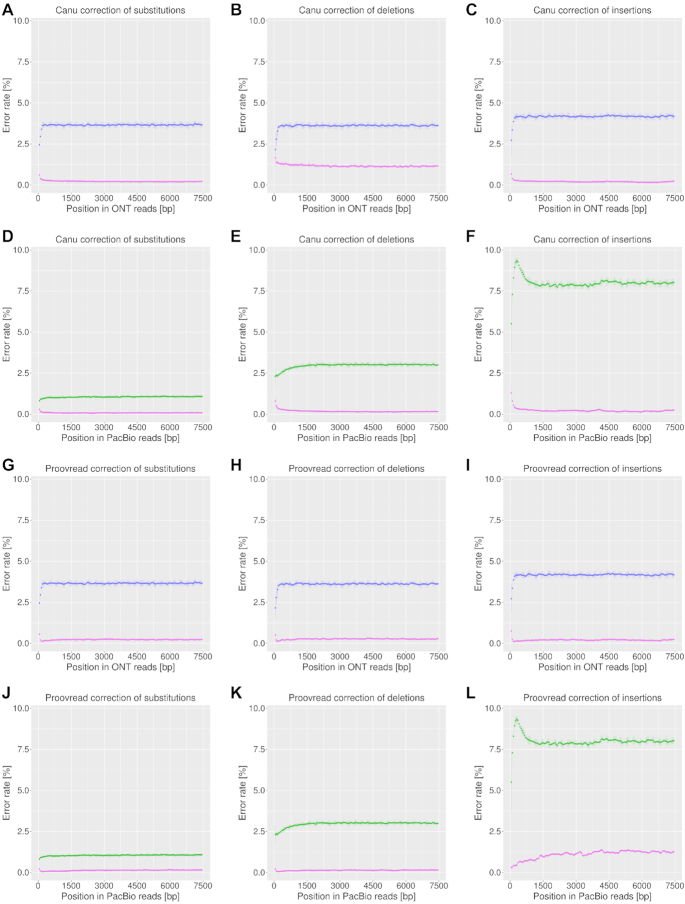
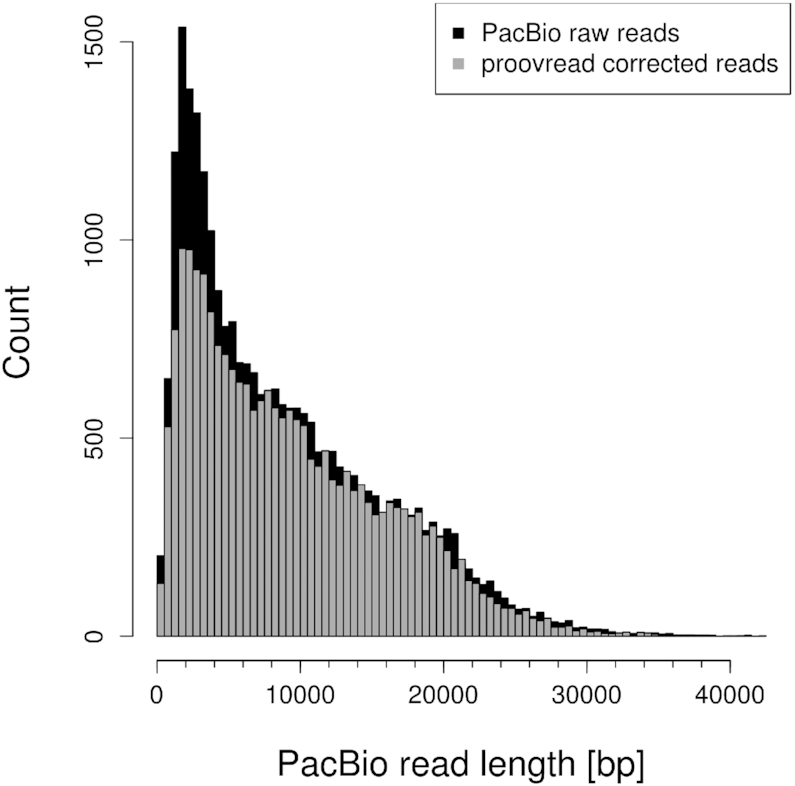
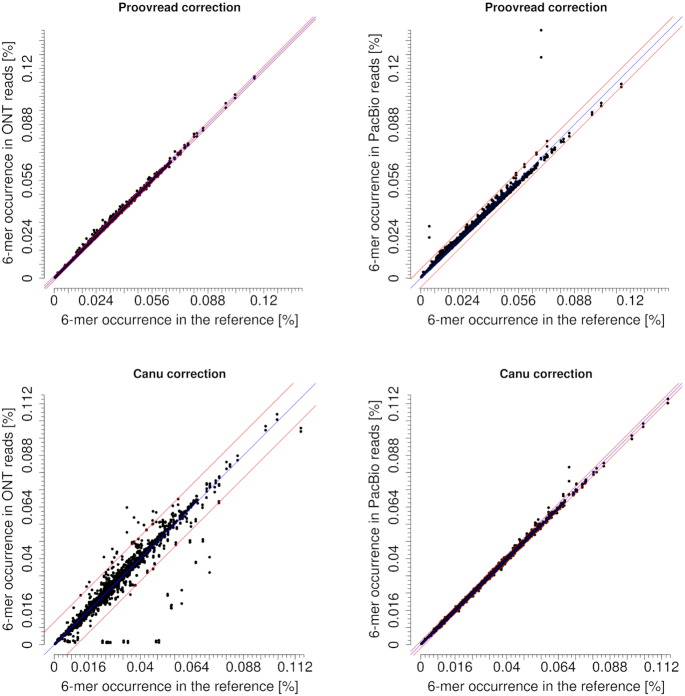
Third-generation sequencing technologies provided by Pacific Biosciences and Oxford Nanopore Technologies generate read lengths in the scale of kilobasepairs. However, these reads display high error rates, and correction steps are necessary to realize their great potential in genomics and transcriptomics. Here, we compare properties of PacBio and Nanopore data and assess correction methods by Canu, MARVEL and proovread in various combinations. We found total error rates of around 13% in the raw datasets. PacBio reads showed a high rate of insertions (around 8%) whereas Nanopore reads showed similar rates for substitutions, insertions and deletions of around 4% each. In data from both technologies the errors were uniformly distributed along reads apart from noisy 5' ends, and homopolymers appeared among the most over-represented kmers relative to a reference. Consensus correction using read overlaps reduced error rates to about 1% when using Canu or MARVEL after patching. The lowest error rate in Nanopore data (0.45%) was achieved by applying proovread on MARVEL-patched data including Illumina short-reads, and the lowest error rate in PacBio data (0.42%) was the result of Canu correction with minimap2 alignment after patching. Our study provides valuable insights and benchmarks regarding long-read data and correction methods.
太平洋生物科学公司(Pacific Biosciences)和牛津纳米孔技术公司(Oxford Nanopore Technologies)提供的第三代测序技术可生成数千碱基对规模的读长。然而,这些读长显示出高错误率,因此需要校正步骤来实现它们在基因组学和转录组学中的巨大潜力。在此,我们比较了PacBio和纳米孔数据的特性,并评估了Canu、MARVEL和proovread以各种组合方式进行的校正方法。我们发现原始数据集中的总错误率约为13%。PacBio读长显示出较高的插入率(约8%),而纳米孔读长在替换、插入和缺失方面的比率相似,均约为4%。在这两种技术的数据中,除了有噪声的5'端外,错误沿读长均匀分布,并且相对于参考序列,同聚物出现在最丰富的kmer之中。当在修补后使用Canu或MARVEL时,利用读长重叠进行的一致性校正可将错误率降低至约1%。通过对包括Illumina短读长在内的MARVEL修补后的数据应用proovread,纳米孔数据实现了最低错误率(0.45%),而PacBio数据的最低错误率(0.42%)是修补后使用minimap2比对进行Canu校正的结果。我们的研究提供了关于长读长数据和校正方法的宝贵见解及基准。