Luu Phuc-Loi, Ong Phuc-Thinh, Dinh Thanh-Phuoc, Clark Susan J
Epigenetics Research Laboratory, Genomics and Epigenetics Division, Garvan Institute of Medical Research, Sydney 2010, New South Wales, Australia.
Faculty of Public Health, University of Medicine and Pharmacy at Ho Chi Minh city, Ho Chi Minh city 70000, Vietnam.
NAR Genom Bioinform. 2020 Aug 6;2(3):lqaa054. doi: 10.1093/nargab/lqaa054. eCollection 2020 Sep.
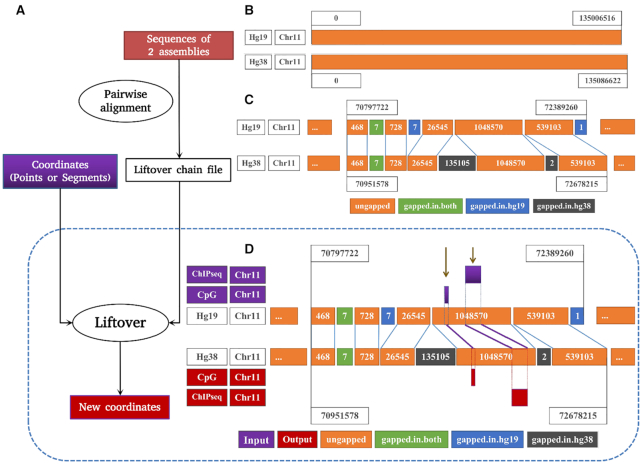
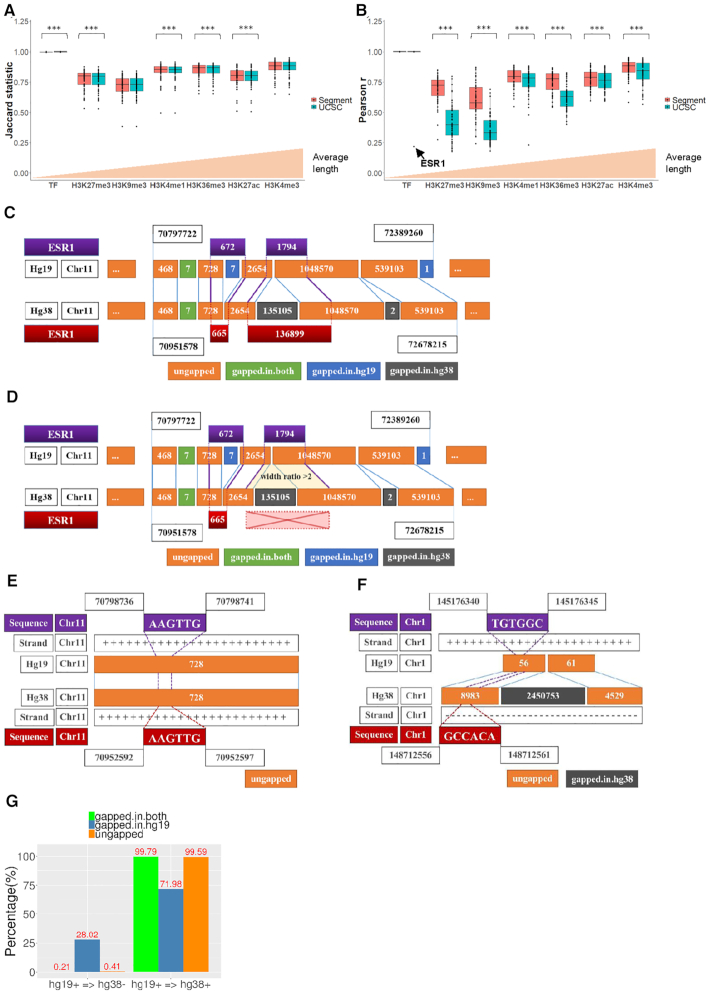
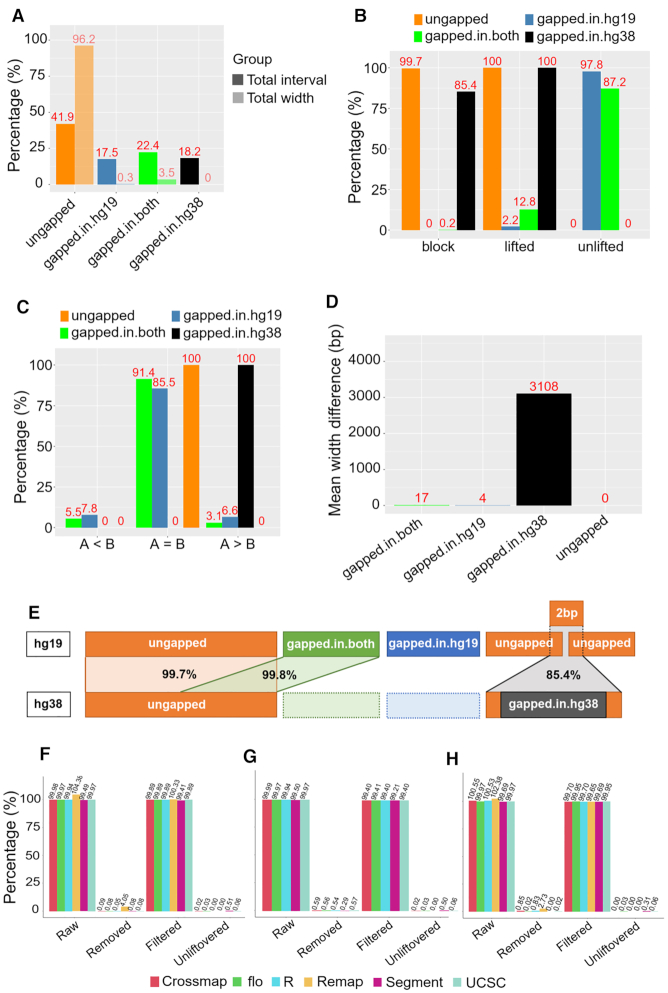
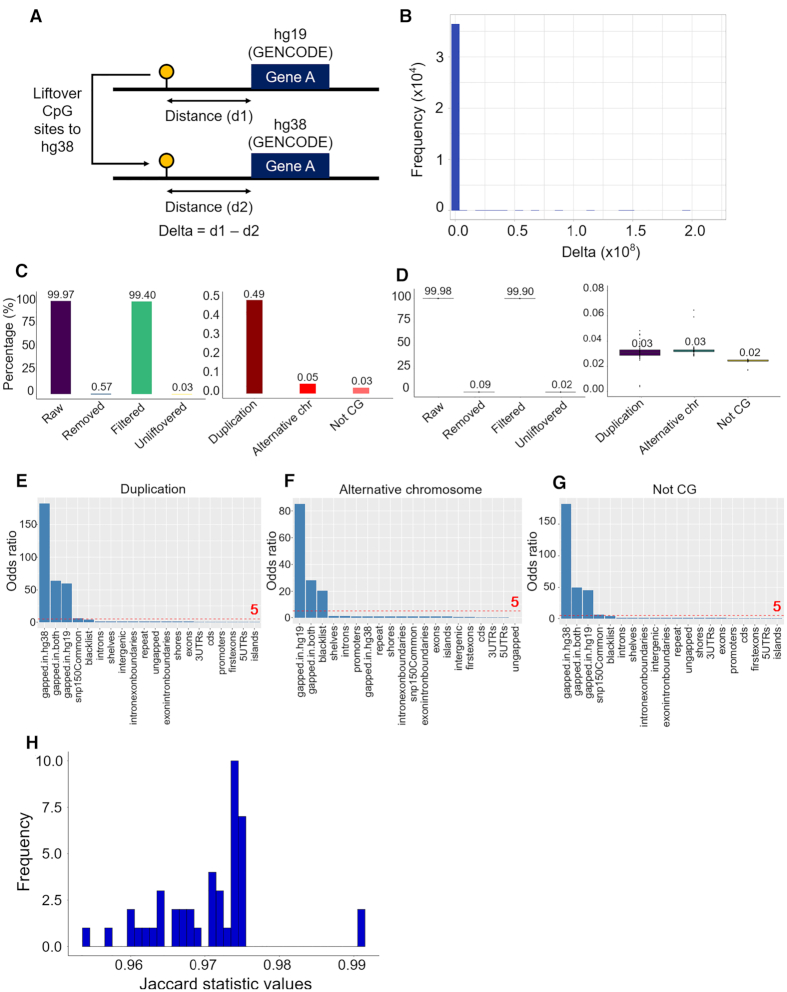
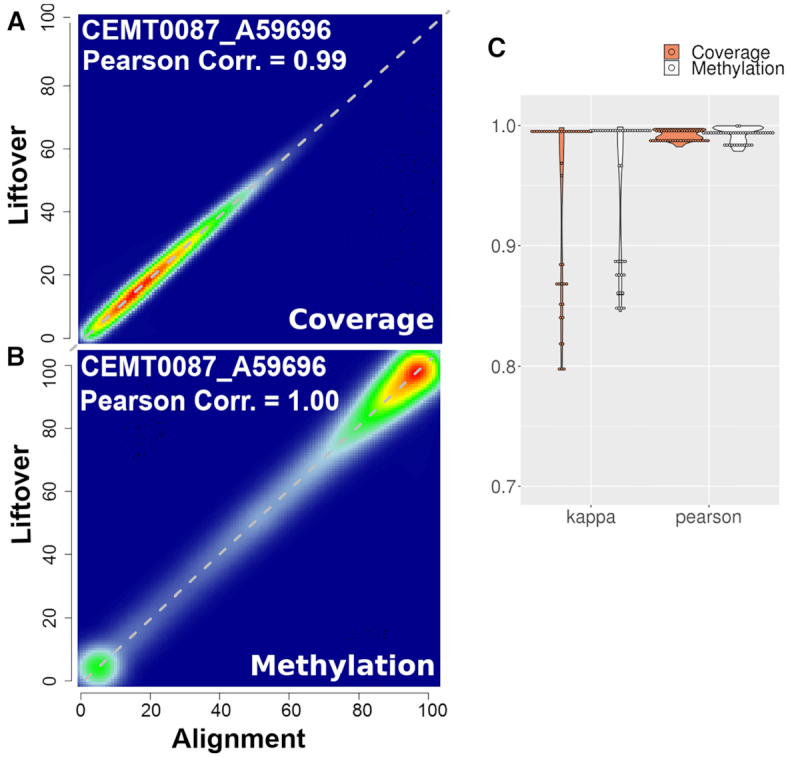
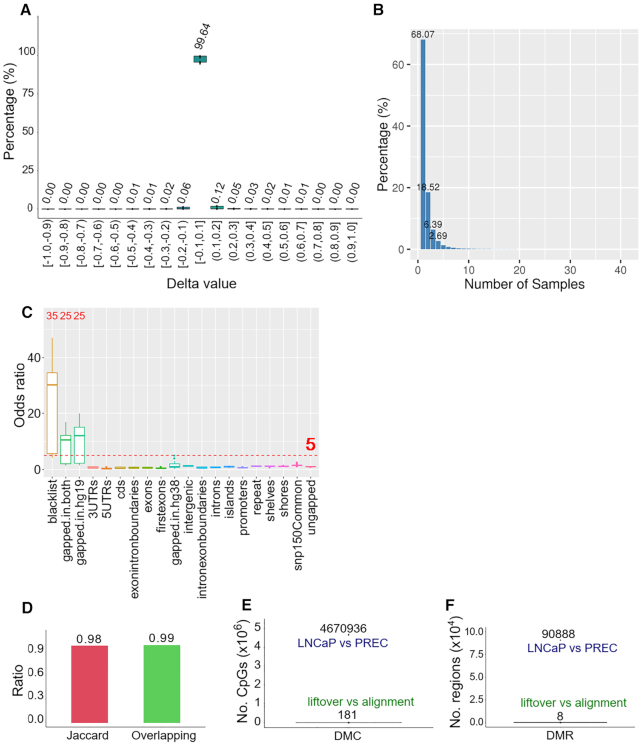
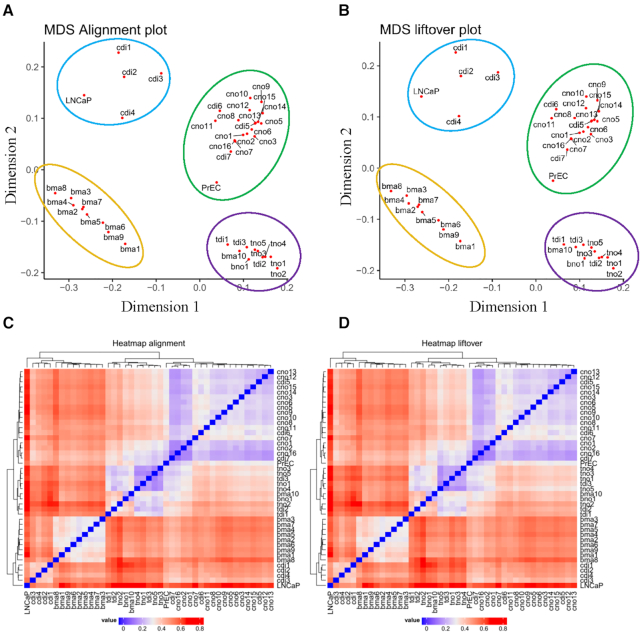
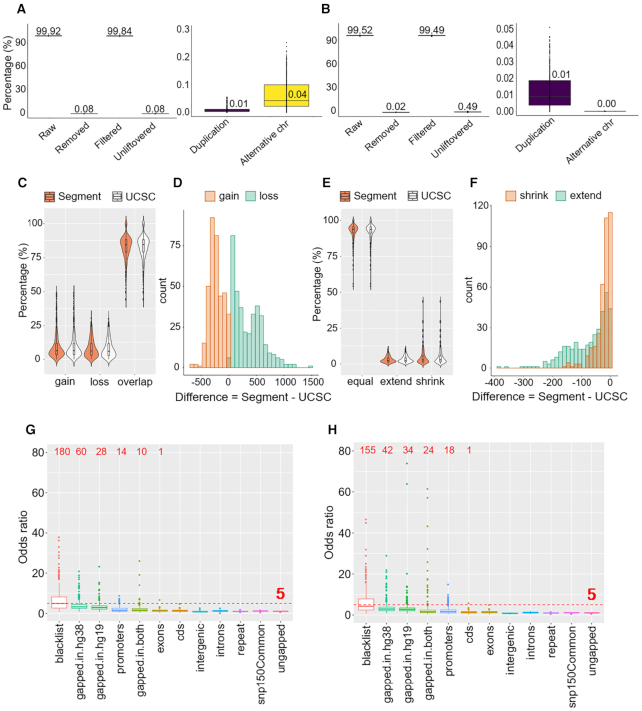
As reference genome assemblies are updated there is a need to convert epigenome sequence data from older genome assemblies to newer versions, to facilitate data integration and visualization on the same coordinate system. Conversion can be done by re-alignment of the original sequence data to the new assembly or by converting the coordinates of the data between assemblies using a mapping file, an approach referred to as 'liftover'. Compared to re-alignment approaches, liftover is a more rapid and cost-effective solution. Here, we benchmark six liftover tools commonly used for conversion between genome assemblies by coordinates, including , , , , and to determine how they performed for whole genome bisulphite sequencing (WGBS) and ChIP-seq data. Our results show high correlation between the six tools for conversion of 43 WGBS paired samples. For the chromatin sequencing data we found from interval conversion of 366 ChIP-Seq datasets, generates more reliable results than . However, we found some regions do not always remain the same after liftover. To further increase the accuracy of liftover and avoid misleading results, we developed a three-step guideline that removes aberrant regions to ensure more robust genome conversion between reference assemblies.
随着参考基因组组装的更新,有必要将表观基因组序列数据从旧的基因组组装版本转换为新的版本,以便于在同一坐标系上进行数据整合和可视化。转换可以通过将原始序列数据重新比对到新的组装版本来完成,或者通过使用映射文件在不同组装版本之间转换数据的坐标来完成,这种方法被称为“坐标转换”。与重新比对方法相比,坐标转换是一种更快速且更具成本效益的解决方案。在这里,我们对六种常用于通过坐标在基因组组装版本之间进行转换的坐标转换工具进行了基准测试,包括 、 、 、 、 和 ,以确定它们在全基因组亚硫酸氢盐测序(WGBS)和染色质免疫沉淀测序(ChIP-seq)数据转换方面的表现。我们的结果表明,在转换43个WGBS配对样本时,这六种工具之间具有高度相关性。对于我们从366个ChIP-Seq数据集的区间转换中获得的染色质测序数据, 生成的结果比 更可靠。然而,我们发现有些区域在坐标转换后并不总是保持不变。为了进一步提高坐标转换的准确性并避免产生误导性结果,我们制定了一个三步指南,用于去除异常区域,以确保在参考组装版本之间进行更稳健的基因组转换。