Li Wan-Chen, Liu Hou-Cheng, Lin Ying-Jyun, Tung Shu-Yun, Wang Ting-Fang
Institute of Molecular Biology, Academia Sinica, Taipei 115, Taiwan.
NAR Genom Bioinform. 2020 Jul 29;2(3):lqaa056. doi: 10.1093/nargab/lqaa056. eCollection 2020 Sep.
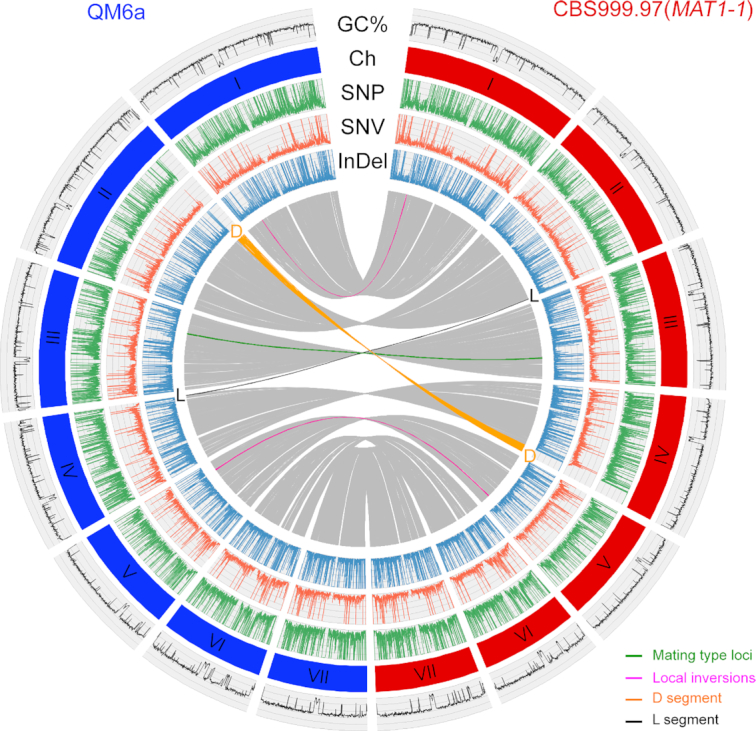
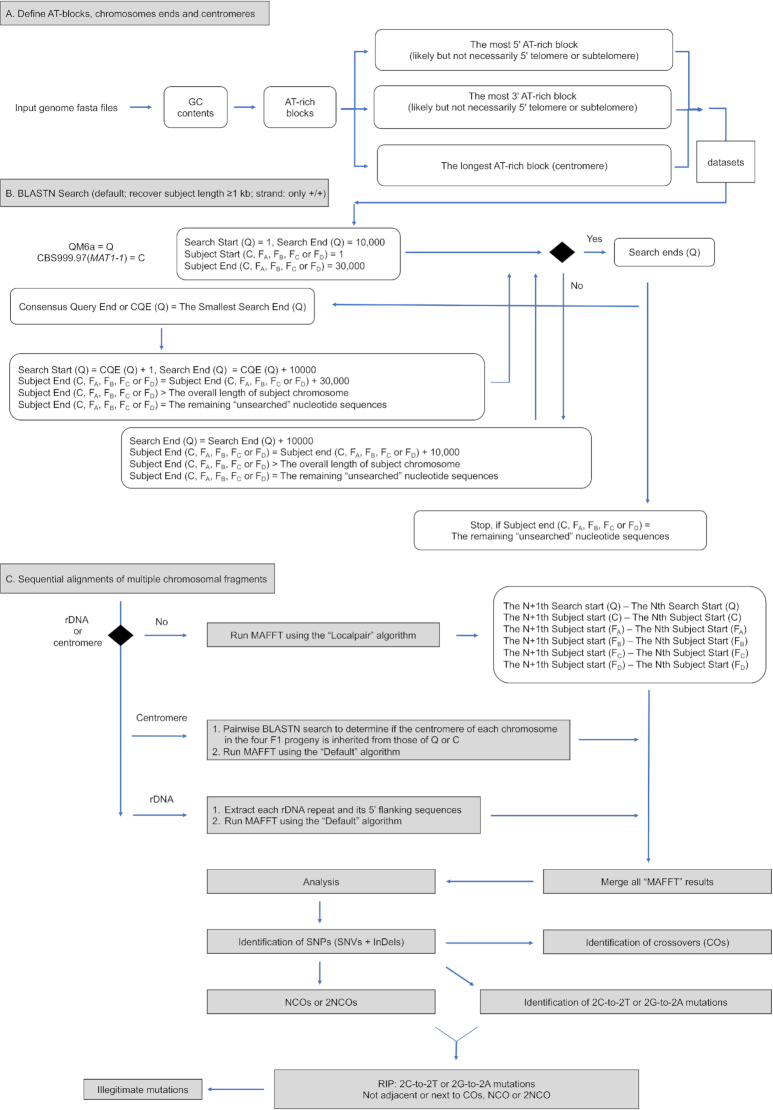
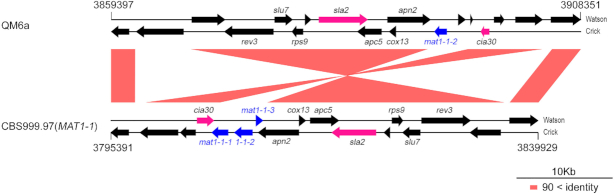
Generation of new genetic diversity by crossover (CO) and non-crossover (NCO) is a fundamental process in eukaryotes. Fungi have played critical roles in studying this process because they permit tetrad analysis, which has been used by geneticists for several decades to determine meiotic recombination products. New genetic variations can also be generated in zygotes via illegitimate mutation (IM) and repeat-induced point mutation (RIP). RIP is a genome defense mechanism for preventing harmful expansion of transposable elements or duplicated sequences in filamentous fungi. Although the exact mechanism of RIP is unknown, the C:G to T:A mutations might result from DNA cytosine methylation. A comprehensive approach for understanding the molecular mechanisms underlying these important processes is to perform high-throughput mapping of CO, NCO, RIP and IM in zygotes bearing large numbers of heterozygous variant markers. To this aim, we developed 'TSETA', a versatile and user-friendly pipeline that utilizes high-quality and chromosome-level genome sequences involved in a single meiotic event of the industrial workhorse fungus . TSETA not only can be applied to most sexual eukaryotes for genome-wide tetrad analysis, it also outcompetes most currently used methods for calling out single nucleotide polymorphisms between two or more intraspecies strains or isolates.
通过交叉(CO)和非交叉(NCO)产生新的遗传多样性是真核生物中的一个基本过程。真菌在研究这一过程中发挥了关键作用,因为它们允许进行四分体分析,几十年来遗传学家一直用这种方法来确定减数分裂重组产物。新的遗传变异也可以通过异常突变(IM)和重复诱导点突变(RIP)在合子中产生。RIP是一种基因组防御机制,用于防止丝状真菌中转座元件或重复序列的有害扩增。虽然RIP的确切机制尚不清楚,但C:G到T:A的突变可能是由DNA胞嘧啶甲基化引起的。理解这些重要过程背后分子机制的一种综合方法是在携带大量杂合变异标记的合子中对CO、NCO、RIP和IM进行高通量定位。为此,我们开发了“TSETA”,这是一个通用且用户友好的流程,它利用了工业主力真菌单个减数分裂事件中涉及的高质量染色体水平基因组序列。TSETA不仅可以应用于大多数有性真核生物进行全基因组四分体分析,它在检测两个或多个种内菌株或分离株之间的单核苷酸多态性方面也优于目前使用的大多数方法。