Yu Kai, Yang Huan, Lv Qiao-Li, Wang Li-Chong, Tan Zi-Long, Zhang Zhe, Ji Yu-Long, Lin Qian-Xia, Chen Jun-Jun, He Wei, Chen Zhen, Shen Xiao-Li
Department of Neurosurgery, The Second Affiliated Hospital of Nanchang University, No. 1 Minde Road, Donghu District, Jiangxi, 330006, Nanchang, People's Republic of China.
Jiangxi Key Laboratory of Translational Cancer Research, Jiangxi Cancer Hospital, Jiangxi, Nanchang, People's Republic of China.
Cancer Cell Int. 2021 Feb 12;21(1):102. doi: 10.1186/s12935-021-01789-z.
Glioblastoma is the most common primary malignant brain tumor. Because of the limited understanding of its pathogenesis, the prognosis of glioblastoma remains poor. This study was conducted to explore potential competing endogenous RNA (ceRNA) network chains and biomarkers in glioblastoma by performing integrated bioinformatics analysis.
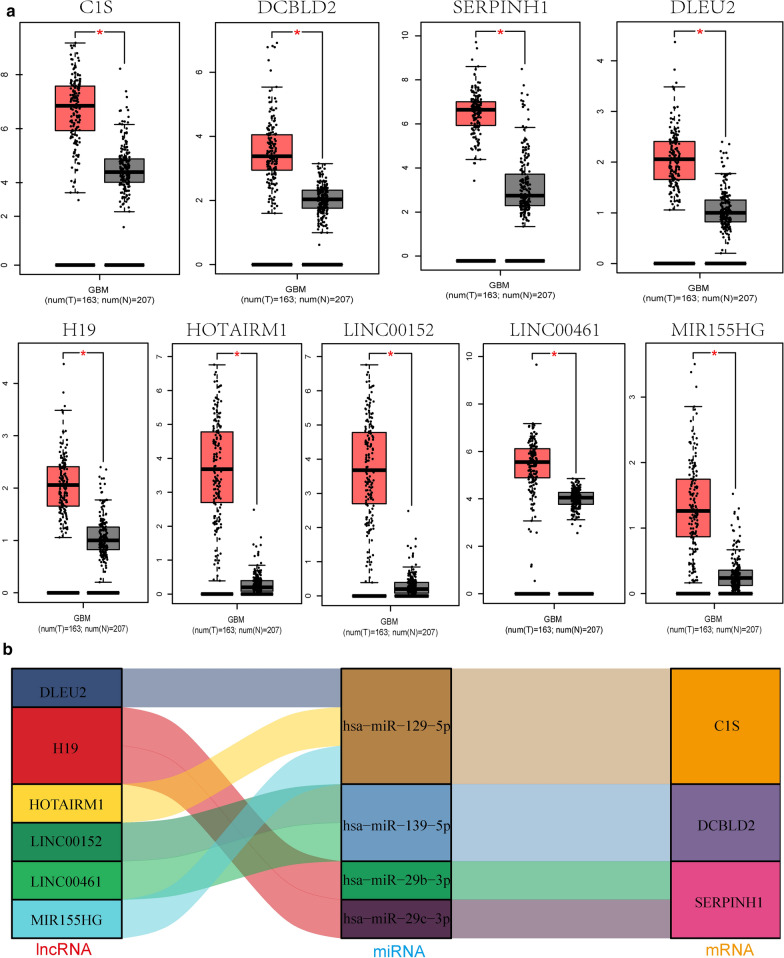
Transcriptome expression data from The Cancer Genome Atlas database and Gene Expression Omnibus were analyzed to identify differentially expressed genes between glioblastoma and normal tissues. Biological pathways potentially associated with the differentially expressed genes were explored by Gene Ontology and Kyoto Encyclopedia of Genes and Genomes pathway analysis, and a protein-protein interaction network was established using the STRING database and Cytoscape. Survival analysis using Gene Expression Profiling Interactive Analysis was based on the Kaplan-Meier curve method. A ceRNA network chain was established using the intersection method to align data from four databases (miRTarBase, miRcode, TargetScan, and lncBace2.0), and expression differences and correlations were verified by quantitative reverse-transcription polymerase chain reaction analysis and by determining the Pearson correlation coefficient. Additionally, an MTS assay and the wound-healing and transwell assays were performed to evaluate the effects of complement C1s (C1S) on the viability and migration and invasion abilities of glioblastoma cells, respectively.
We detected 2842 differentially expressed (DE) mRNAs, 2577 DE long non-coding RNAs (lncRNAs), and 309 DE microRNAs (miRNAs) that were dysregulated in glioblastoma. The final ceRNA network consisted of six specific lncRNAs, four miRNAs, and four mRNAs. Among them, four DE mRNAs and one DE lncRNA were correlated with overall survival (p < 0.05). C1S was significantly correlated with overall survival (p= 0.015). In functional assays, knockdown of C1S inhibited the proliferation and invasion of glioblastoma cell lines.
We established four ceRNA networks that may influence the occurrence and development of glioblastoma. Among them, the MIR155HG/has-miR-129-5p/C1S axis is a potential marker and therapeutic target for glioblastoma. Knockdown of C1S inhibited the proliferation, migration, and invasion of glioblastoma cells. These findings clarify the role of the ceRNA regulatory network in glioblastoma and provide a foundation for further research.
胶质母细胞瘤是最常见的原发性恶性脑肿瘤。由于对其发病机制的了解有限,胶质母细胞瘤的预后仍然很差。本研究旨在通过综合生物信息学分析,探索胶质母细胞瘤中潜在的竞争性内源性RNA(ceRNA)网络链和生物标志物。
分析来自癌症基因组图谱数据库和基因表达综合数据库的转录组表达数据,以鉴定胶质母细胞瘤与正常组织之间差异表达的基因。通过基因本体论和京都基因与基因组百科全书通路分析,探索与差异表达基因潜在相关的生物学通路,并使用STRING数据库和Cytoscape建立蛋白质-蛋白质相互作用网络。使用基因表达谱交互式分析进行的生存分析基于Kaplan-Meier曲线法。使用交集法建立ceRNA网络链,以对齐来自四个数据库(miRTarBase、miRcode、TargetScan和lncBace2.0)的数据,并通过定量逆转录聚合酶链反应分析和确定Pearson相关系数来验证表达差异和相关性。此外,进行MTS试验以及伤口愈合试验和Transwell试验,分别评估补体C1s(C1S)对胶质母细胞瘤细胞活力、迁移和侵袭能力的影响。
我们检测到在胶质母细胞瘤中失调的2842个差异表达(DE)mRNA、2577个DE长链非编码RNA(lncRNA)和309个DE微小RNA(miRNA)。最终的ceRNA网络由六个特定的lncRNA、四个miRNA和四个mRNA组成。其中,四个DE mRNA和一个DE lncRNA与总生存期相关(p<0.05)。C1S与总生存期显著相关(p=0.015)。在功能试验中,敲低C1S可抑制胶质母细胞瘤细胞系的增殖和侵袭。
我们建立了四个可能影响胶质母细胞瘤发生和发展的ceRNA网络。其中,MIR155HG/has-miR-129-5p/C1S轴是胶质母细胞瘤的潜在标志物和治疗靶点。敲低C1S可抑制胶质母细胞瘤细胞的增殖、迁移和侵袭。这些发现阐明了ceRNA调控网络在胶质母细胞瘤中的作用,并为进一步研究提供了基础。