Eswaramoorthy Rajalakshmanan, Hailekiros Hadgu, Kedir Fedlu, Endale Milkyas
Department of Applied Chemistry, School of Applied Natural Science, Adama Science and Technology University, Adama, 1888, Ethiopia.
Adv Appl Bioinform Chem. 2021 Feb 5;14:13-24. doi: 10.2147/AABC.S290912. eCollection 2021.
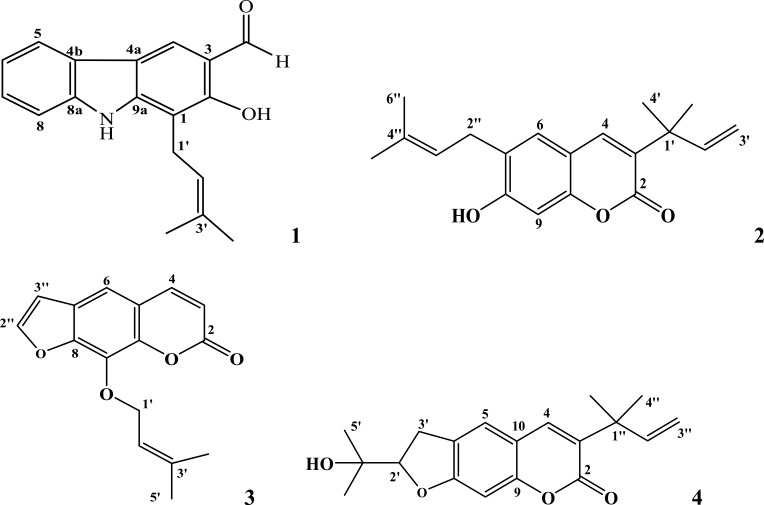
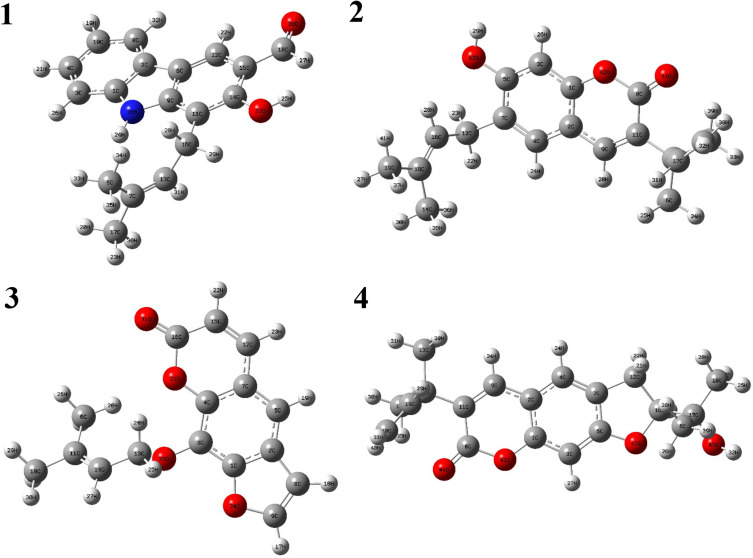
In modern drug design, in silico methods are largely used to understand drug-receptor interactions and quantum chemical properties. In the present study, a computational de novo design approach was used to confirm mode of binding for antibacterial activity, elucidating quantum chemical properties and ADMET-drug-likeness of carbazole alkaloid () and three coumarins (-) isolated from roots of .
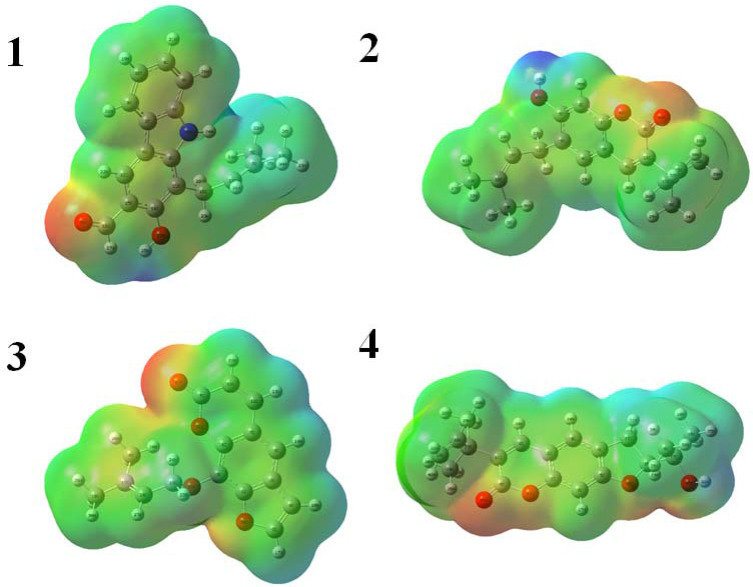
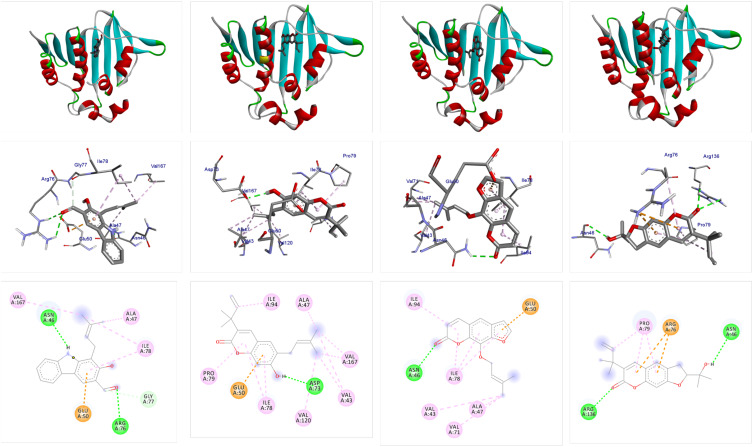
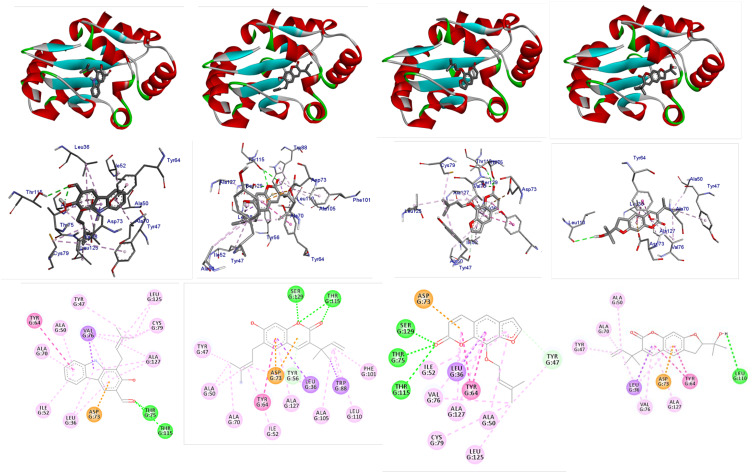
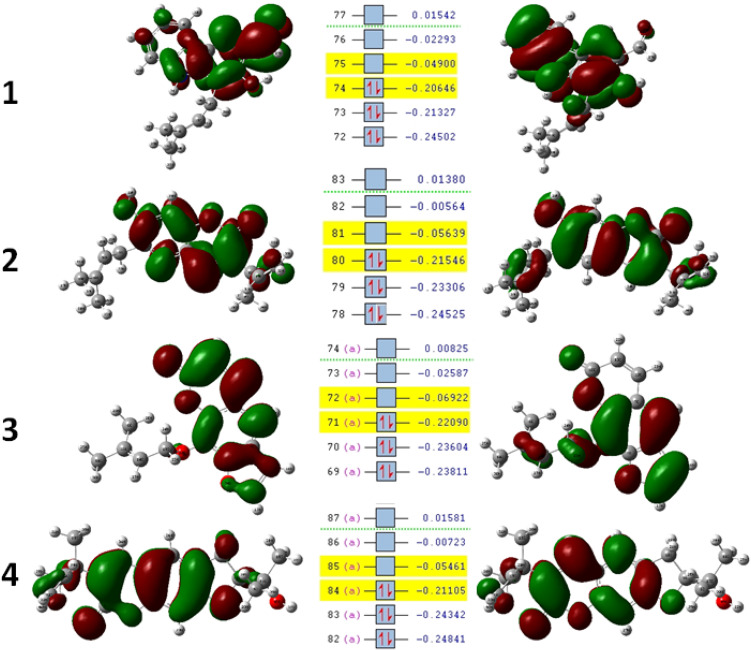
Docking studies were performed with DNA-Gyrase (6F86) and LasR binding domain (2UV0) employing a flexible ligand docking approach using AutoDock Vina. SwissADME prediction and toxicological predictions were performed by ADMET. The optimized structures and molecular electrostatic potential surface of the isolated compounds were predicted by DFT analysis using B3LYP/6-31G basis levels.
The docking results revealed that compound showed better docking scores against both DNA gyrase B and LasR binding domain compared with ciprofloxacin with potential as an inhibitor of bacterial DNA gyrase and quorum sensing LasR binding domain. The SwissADME prediction results showed that all the isolated compounds () satisfy Lipinski's rule of five with zero violations. Toxicological prediction results suggested that all compounds and ciprofloxacin are non-hepatotoxic, non-carcinogenic, non-irritant, immunogenic, and non-cytotoxic. The DFT analysis results revealed that compound has large electronegativity (χeV), global softness (σ eV), global electrophilicity (ωeV), and mutagenicity value closer to ciprofloxacin (with LD value of 480 mg/kg) suggesting better bioactivity and chemical reactivity with considerable intra-molecular charge transfer between electron-donor to electron-acceptor groups.
Overall, compound may serve as a lead molecule that could be developed into a potent DNA gyrase B inhibitor and efficient inhibitor for quorum sensing auto-inducer LasR binding domain of .
在现代药物设计中,计算机模拟方法在很大程度上用于理解药物与受体的相互作用以及量子化学性质。在本研究中,采用计算从头设计方法来确认咔唑生物碱()和从的根部分离出的三种香豆素(-)的抗菌活性结合模式,阐明其量子化学性质和ADMET药物相似性。
使用AutoDock Vina的柔性配体对接方法,对DNA解旋酶(6F86)和LasR结合结构域(2UV0)进行对接研究。通过ADMET进行SwissADME预测和毒理学预测。使用B3LYP/6-31G基组水平的DFT分析预测分离化合物的优化结构和分子静电势表面。
对接结果表明,与环丙沙星相比,化合物对DNA解旋酶B和LasR结合结构域均显示出更好的对接分数,具有作为细菌DNA解旋酶抑制剂和群体感应LasR结合结构域抑制剂的潜力。SwissADME预测结果表明,所有分离的化合物()均满足Lipinski的五规则,无违规情况。毒理学预测结果表明,所有化合物和环丙沙星均无肝毒性、无致癌性、无刺激性、无免疫原性且无细胞毒性。DFT分析结果表明,化合物具有较大的电负性(χeV)、全局软度(σ eV)、全局亲电性(ωeV),且致突变性值接近环丙沙星(LD值为480 mg/kg),表明其具有更好的生物活性和化学反应性,在电子供体基团与电子受体基团之间存在相当程度的分子内电荷转移。
总体而言,化合物可作为先导分子,有望开发成为一种有效的DNA解旋酶B抑制剂和群体感应自诱导物LasR结合结构域的高效抑制剂。