Tang Xiaolu, Ying Ruochen, Yao Xinmin, Li Guanghao, Wu Changcheng, Tang Yiyuli, Li Zhida, Kuang Bishan, Wu Feng, Chi Changsheng, Du Xiaoman, Qin Yi, Gao Shenghan, Hu Songnian, Ma Juncai, Liu Tiangang, Pang Xinghuo, Wang Jianwei, Zhao Guoping, Tan Wenjie, Zhang Yaping, Lu Xuemei, Lu Jian
State Key Laboratory of Protein and Plant Gene Research, Center for Bioinformatics, School of Life Sciences, Peking University, Beijing 100871, China.
State Key Laboratory of Genetic Resources and Evolution, Kunming Institute of Zoology, Center for Excellence in Animal Evolution and Genetics, Chinese Academy of Sciences, Kunming 650223, China.
Sci Bull (Beijing). 2021 Nov 30;66(22):2297-2311. doi: 10.1016/j.scib.2021.02.012. Epub 2021 Feb 6.
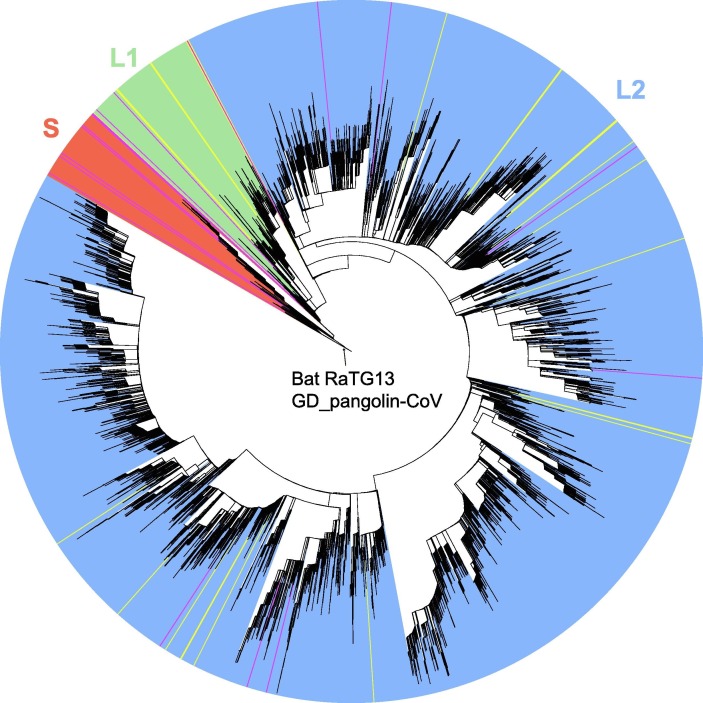
The pandemic due to the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), the etiological agent of coronavirus disease 2019 (COVID-19), has caused immense global disruption. With the rapid accumulation of SARS-CoV-2 genome sequences, however, thousands of genomic variants of SARS-CoV-2 are now publicly available. To improve the tracing of the viral genomes' evolution during the development of the pandemic, we analyzed single nucleotide variants (SNVs) in 121,618 high-quality SARS-CoV-2 genomes. We divided these viral genomes into two major lineages (L and S) based on variants at sites 8782 and 28144, and further divided the L lineage into two major sublineages (L1 and L2) using SNVs at sites 3037, 14408, and 23403. Subsequently, we categorized them into 130 sublineages (37 in S, 35 in L1, and 58 in L2) based on marker SNVs at 201 additional genomic sites. This lineage/sublineage designation system has a hierarchical structure and reflects the relatedness among the subclades of the major lineages. We also provide a companion website (www.covid19evolution.net) that allows users to visualize sublineage information and upload their own SARS-CoV-2 genomes for sublineage classification. Finally, we discussed the possible roles of compensatory mutations and natural selection during SARS-CoV-2's evolution. These efforts will improve our understanding of the temporal and spatial dynamics of SARS-CoV-2's genome evolution.
由严重急性呼吸综合征冠状病毒2(SARS-CoV-2)引起的大流行,即2019冠状病毒病(COVID-19)的病原体,已在全球造成巨大破坏。然而,随着SARS-CoV-2基因组序列的迅速积累,现在已有数千个SARS-CoV-2基因组变体公开可用。为了更好地追踪大流行发展过程中病毒基因组的进化,我们分析了121618个高质量SARS-CoV-2基因组中的单核苷酸变体(SNV)。我们根据8782和28144位点的变体将这些病毒基因组分为两个主要谱系(L和S),并使用3037、14408和23403位点的SNV将L谱系进一步分为两个主要亚谱系(L1和L2)。随后,我们根据另外201个基因组位点的标记SNV将它们分为130个亚谱系(S中有37个,L1中有35个,L2中有58个)。这种谱系/亚谱系指定系统具有层次结构,反映了主要谱系的亚分支之间的相关性。我们还提供了一个配套网站(www.covid19evolution.net),用户可以在该网站上可视化亚谱系信息,并上传自己的SARS-CoV-2基因组进行亚谱系分类。最后,我们讨论了SARS-CoV-2进化过程中补偿性突变和自然选择可能发挥的作用。这些努力将增进我们对SARS-CoV-2基因组进化的时空动态的理解。