McMullen Calum J, Chalmers Susan, Wood Rachel, Cunningham Margaret R, Currie Susan
Strathclyde Institute of Pharmacy and Biomedical Sciences, University of Strathclyde, Glasgow, United Kingdom.
Front Cardiovasc Med. 2021 Feb 1;7:630480. doi: 10.3389/fcvm.2020.630480. eCollection 2020.
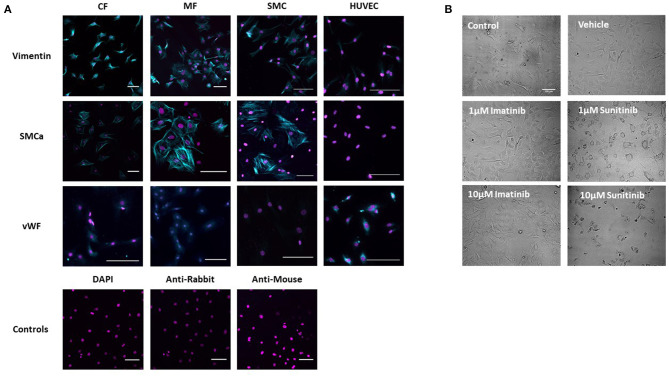
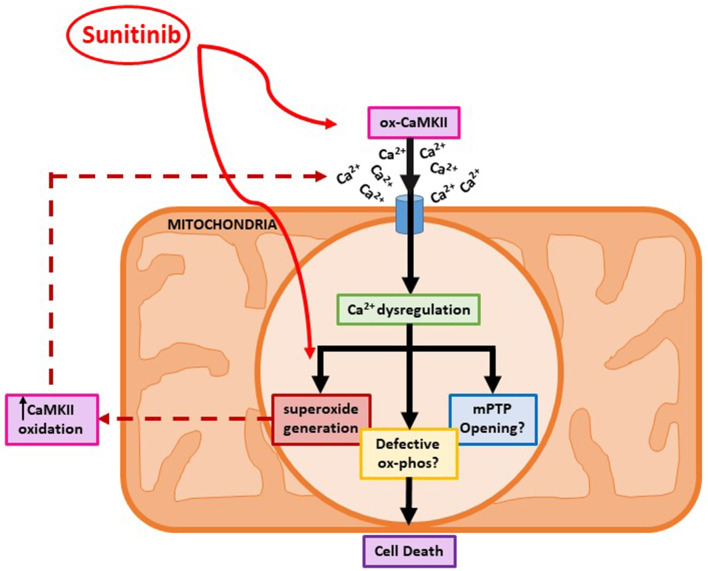
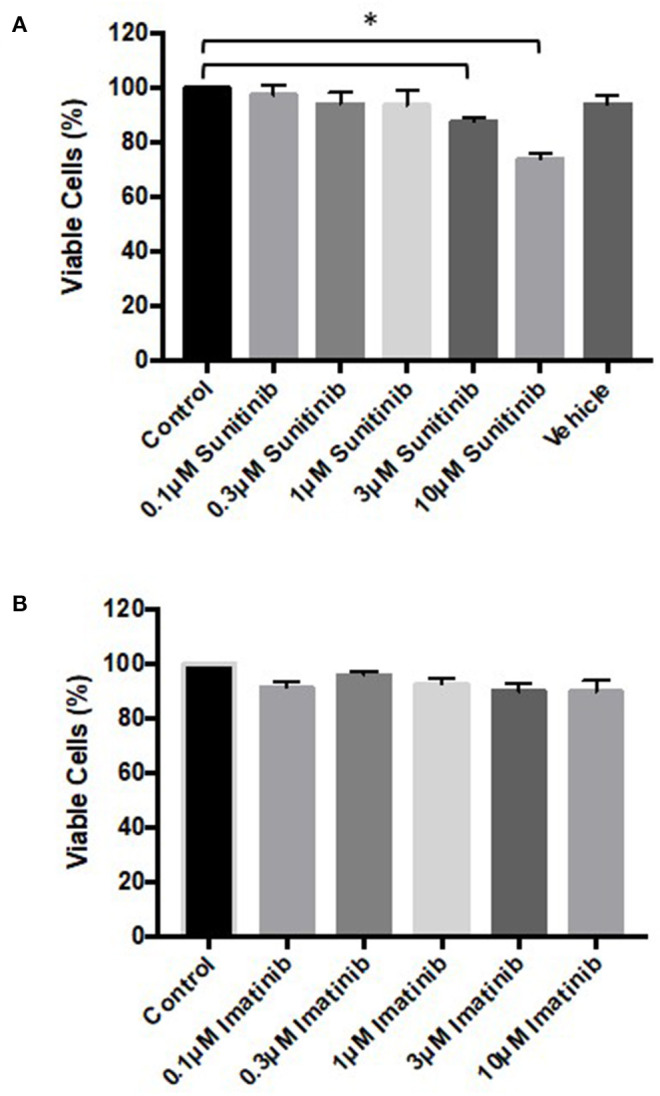
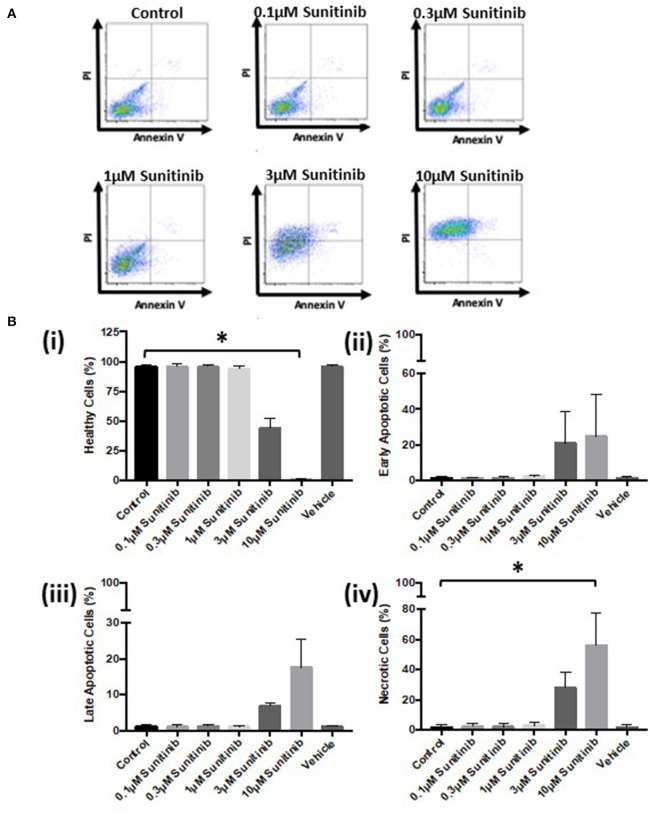
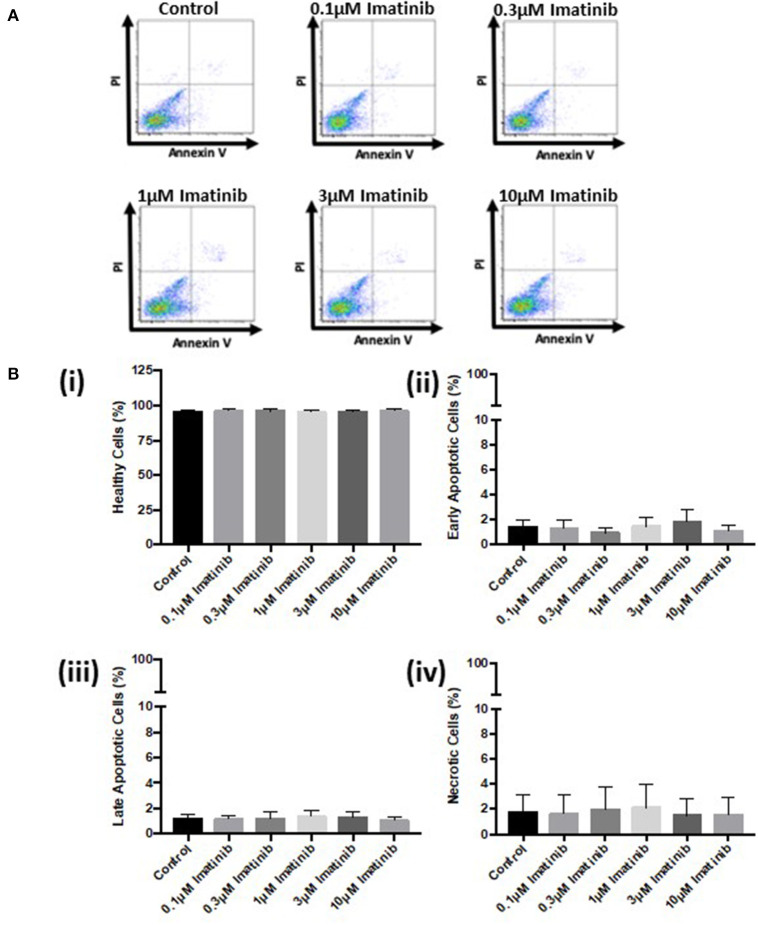
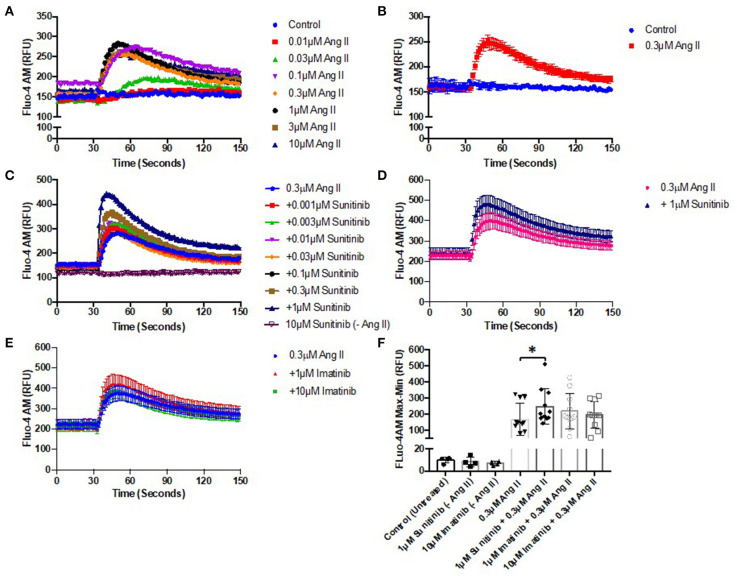
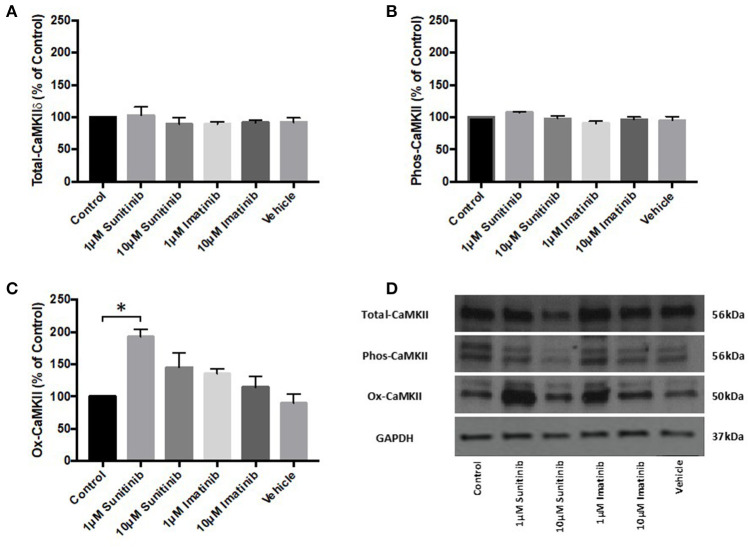
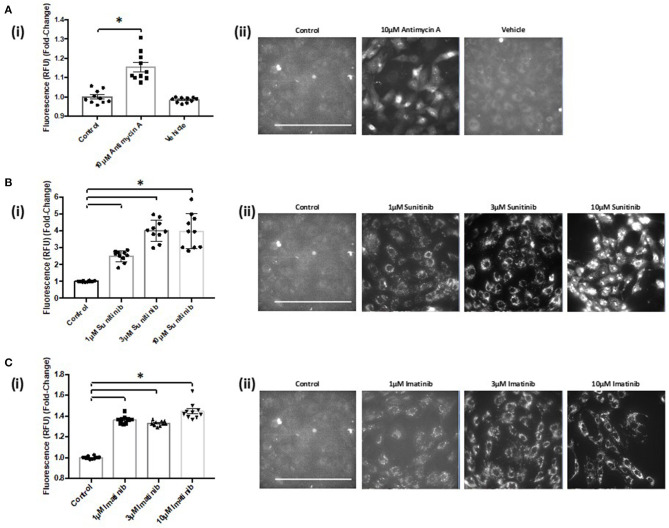
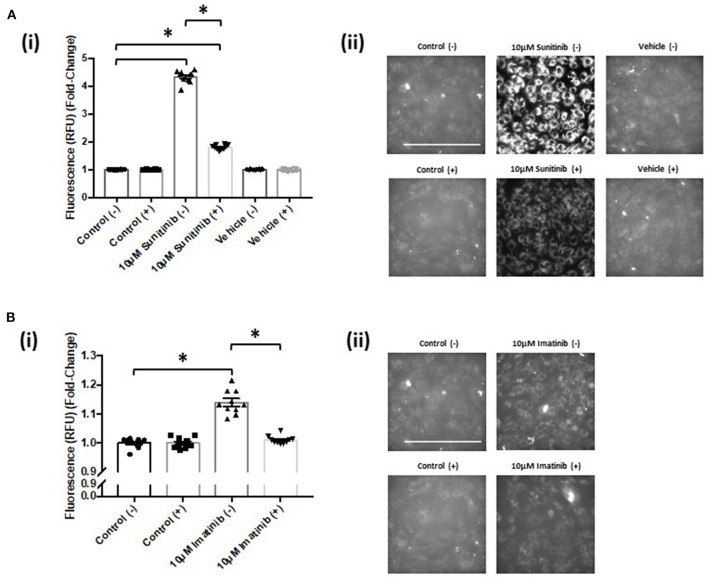
Tyrosine kinase inhibitors (TKIs) have dramatically improved cancer treatment but are known to cause cardiotoxicity. The pathophysiological consequences of TKI therapy are likely to manifest across different cell types of the heart, yet there is little understanding of the differential adverse cellular effects. Cardiac fibroblasts (CFs) play a pivotal role in the repair and remodeling of the heart following insult or injury, yet their involvement in anti-cancer drug induced cardiotoxicity has been largely overlooked. Here, we examine the direct effects of sunitinib malate and imatinib mesylate on adult rat CF viability, Ca handling and mitochondrial function that may contribute to TKI-induced cardiotoxicity. In particular, we investigate whether Ca/calmodulin dependent protein kinase II (CaMKII), may be a mediator of TKI-induced effects. CF viability in response to chronic treatment with both drugs was assessed using MTT assays and flow cytometry analysis. Calcium mobilization was assessed in CFs loaded with Fluo4-AM and CaMKII activation oxidation was measured quantitative immunoblotting. Effects of both drugs on mitochondrial function was determined by live mitochondrial imaging using MitoSOX red. Treatment of CFs with sunitinib (0.1-10 μM) resulted in concentration-dependent alterations in CF phenotype, with progressively significant cell loss at higher concentrations. Flow cytometry analysis and MTT assays revealed increased cell apoptosis and necrosis with increasing concentrations of sunitinib. In contrast, equivalent concentrations of imatinib resulted in no significant change in cell viability. Both sunitinib and imatinib pre-treatment increased Angiotensin II-induced intracellular Ca mobilization, with only sunitinib resulting in a significant effect and also causing increased CaMKII activation oxidation. Live cell mitochondrial imaging using MitoSOX red revealed that both sunitinib and imatinib increased mitochondrial superoxide production in a concentration-dependent manner. This effect in response to both drugs was suppressed in the presence of the CaMKII inhibitor KN-93. Sunitinib and imatinib showed differential effects on CFs, with sunitinib causing marked changes in cell viability at concentrations where imatinib had no effect. Sunitinib caused a significant increase in Angiotensin II-induced intracellular Ca mobilization and both TKIs caused increased mitochondrial superoxide production. Targeted CaMKII inhibition reversed the TKI-induced mitochondrial damage. These findings highlight a new role for CaMKII in TKI-induced cardiotoxicity, particularly at the level of the mitochondria, and confirm differential off-target toxicity in CFs, consistent with the differential selectivity of sunitinib and imatinib.
酪氨酸激酶抑制剂(TKIs)显著改善了癌症治疗,但已知会引起心脏毒性。TKI治疗的病理生理后果可能在心脏的不同细胞类型中表现出来,但对不同的不良细胞效应了解甚少。心脏成纤维细胞(CFs)在心脏受到损伤或伤害后的修复和重塑中起关键作用,然而它们在抗癌药物诱导的心脏毒性中的作用在很大程度上被忽视了。在这里,我们研究了苹果酸舒尼替尼和甲磺酸伊马替尼对成年大鼠CF活力、钙处理和线粒体功能的直接影响,这些影响可能导致TKI诱导的心脏毒性。特别是,我们研究了钙/钙调蛋白依赖性蛋白激酶II(CaMKII)是否可能是TKI诱导效应的介质。使用MTT试验和流式细胞术分析评估了两种药物长期处理后CF的活力。在加载Fluo4-AM的CF中评估钙动员,并通过定量免疫印迹测量CaMKII激活和氧化。使用MitoSOX red进行实时线粒体成像确定了两种药物对线粒体功能的影响。用舒尼替尼(0.1-10μM)处理CF导致CF表型发生浓度依赖性改变,在较高浓度下细胞损失逐渐显著。流式细胞术分析和MTT试验显示,随着舒尼替尼浓度的增加,细胞凋亡和坏死增加。相比之下,同等浓度的伊马替尼导致细胞活力无显著变化。舒尼替尼和伊马替尼预处理均增加了血管紧张素II诱导的细胞内钙动员,只有舒尼替尼产生显著影响,还导致CaMKII激活和氧化增加。使用MitoSOX red进行活细胞线粒体成像显示,舒尼替尼和伊马替尼均以浓度依赖性方式增加线粒体超氧化物的产生。在存在CaMKII抑制剂KN-93的情况下,对两种药物的这种反应受到抑制。舒尼替尼和伊马替尼对CFs显示出不同的影响,舒尼替尼在伊马替尼无作用的浓度下导致细胞活力发生显著变化。舒尼替尼导致血管紧张素II诱导的细胞内钙动员显著增加,两种TKIs均导致线粒体超氧化物产生增加。靶向CaMKII抑制可逆转TKI诱导的线粒体损伤。这些发现突出了CaMKII在TKI诱导的心脏毒性中的新作用,特别是在线粒体水平,并证实了CFs中的不同脱靶毒性,这与舒尼替尼和伊马替尼的不同选择性一致。