Department of Biomedical Informatics, University of Arkansas for Medical Sciences, 4301 W Markham St, Little Rock, AR, 72205, USA.
BMC Bioinformatics. 2021 Feb 18;22(1):77. doi: 10.1186/s12859-021-03995-y.
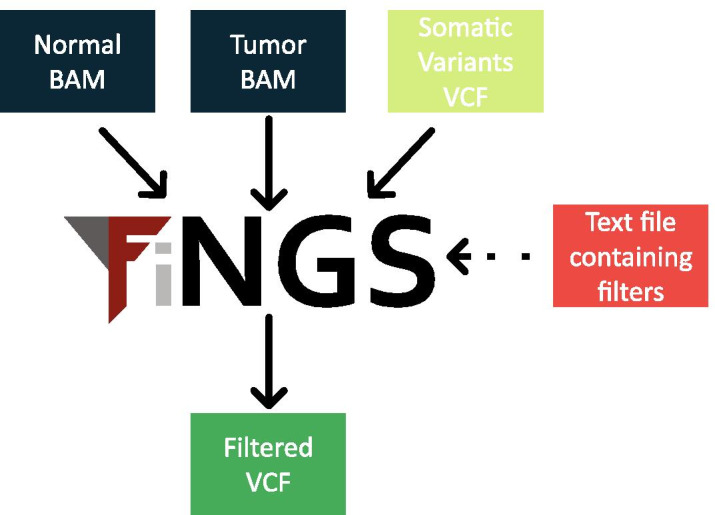
Somatic variant callers are used to find mutations in sequencing data from cancer samples. They are very sensitive and have high recall, but also may produce low precision data with a large proportion of false positives. Further ad hoc filtering is commonly performed after variant calling and before further analysis. Improving the filtering of somatic variants in a reproducible way represents an unmet need. We have developed Filters for Next Generation Sequencing (FiNGS), software written specifically to address these filtering issues.
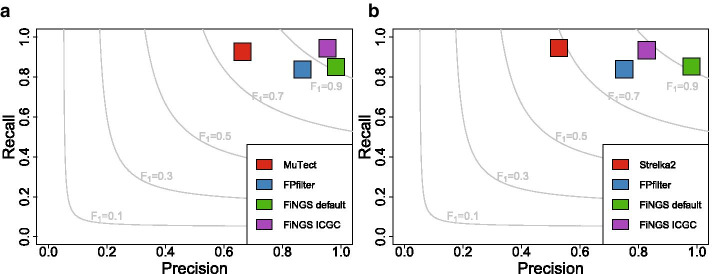
Developed and tested using publicly available sequencing data sets, we demonstrate that FiNGS reliably improves upon the precision of default variant caller outputs and performs better than other tools designed for the same task.
FiNGS provides researchers with a tool to reproducibly filter somatic variants that is simple to both deploy and use, with filters and thresholds that are fully configurable by the user. It ingests and emits standard variant call format (VCF) files and will slot into existing sequencing pipelines. It allows users to develop and implement their own filtering strategies and simple sharing of these with others.
体细胞变异调用程序用于在癌症样本的测序数据中寻找突变。它们非常敏感,召回率高,但也可能产生大量假阳性的低精度数据。在变体调用后和进一步分析之前,通常会进行额外的特定于任务的过滤。以可重复的方式改进体细胞变异的过滤是一个未满足的需求。我们开发了用于下一代测序的过滤器(FiNGS),这是专门为解决这些过滤问题而编写的软件。
使用公开可用的测序数据集进行开发和测试,我们证明 FiNGS 可靠地提高了默认变体调用程序输出的精度,并且比其他专为同一任务设计的工具表现更好。
FiNGS 为研究人员提供了一种可重复过滤体细胞变异的工具,它易于部署和使用,用户可以完全配置过滤器和阈值。它可以接收和输出标准变异调用格式(VCF)文件,并适用于现有的测序管道。它允许用户开发和实施自己的过滤策略,并与他人简单地共享这些策略。