Jernigan Robert L, Sankar Kannan, Jia Kejue, Faraggi Eshel, Kloczkowski Andrzej
Roy J. Carver Department of Biochemistry, Biophysics and Molecular Biology, Iowa State University, Ames, IA, United States.
Research and Information Systems, LLC, Indianapolis, IN, United States.
Front Mol Biosci. 2021 Feb 3;7:607323. doi: 10.3389/fmolb.2020.607323. eCollection 2020.
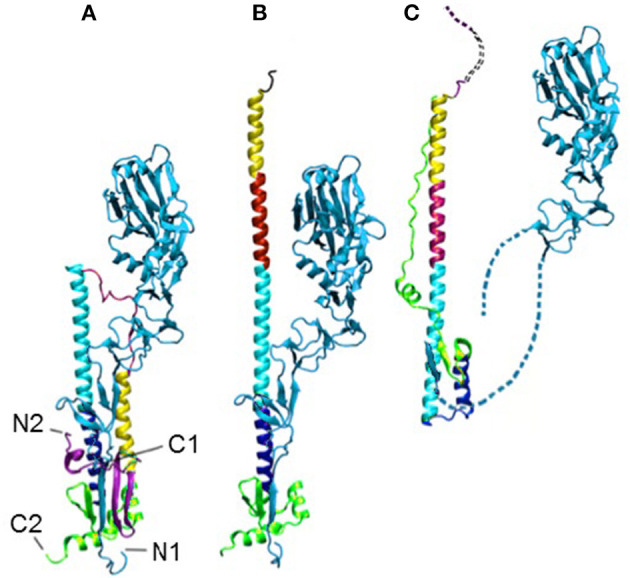
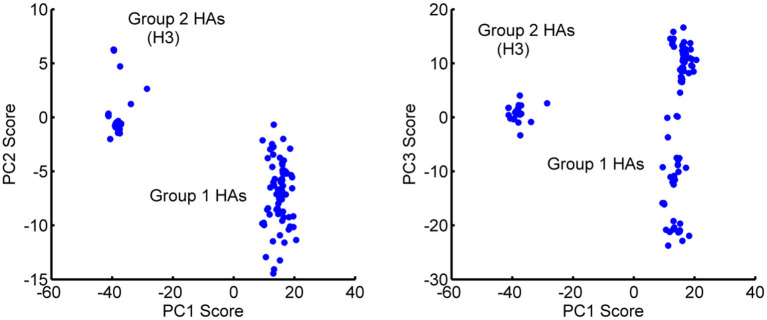
Two new computational approaches are described to aid in the design of new peptide-based drugs by evaluating ensembles of protein structures from their dynamics and through the assessing of structures using empirical contact potential. These approaches build on the concept that conformational variability can aid in the binding process and, for disordered proteins, can even facilitate the binding of more diverse ligands. This latter consideration indicates that such a design process should be less restrictive so that multiple inhibitors might be effective. The example chosen here focuses on proteins/peptides that bind to hemagglutinin (HA) to block the large-scale conformational change for activation. Variability in the conformations is considered from sets of experimental structures, or as an alternative, from their simple computed dynamics; the set of designe peptides/small proteins from the David Baker lab designed to bind to hemagglutinin, is the large set considered and is assessed with the new empirical contact potentials.
本文描述了两种新的计算方法,通过从蛋白质结构动力学评估其结构集合,并使用经验接触势评估结构,来辅助设计基于肽的新型药物。这些方法基于这样的概念,即构象变异性有助于结合过程,对于无序蛋白质,甚至可以促进更多样化配体的结合。后一种考虑表明,这样的设计过程应该限制较少,以便多种抑制剂可能有效。这里选择的例子集中在与血凝素(HA)结合以阻断激活的大规模构象变化的蛋白质/肽。构象的变异性是从实验结构集合中考虑的,或者作为替代方案,从其简单计算的动力学中考虑;来自大卫·贝克实验室的旨在与血凝素结合的设计肽/小蛋白质集合,是所考虑的大集合,并使用新的经验接触势进行评估。