Peng Chao, Huang Yu, Bian Chao, Li Jia, Liu Jie, Zhang Kai, You Xinxin, Lin Zhilong, He Yanbin, Chen Jieming, Lv Yunyun, Ruan Zhiqiang, Zhang Xinhui, Yi Yunhai, Li Yanping, Lin Xueqiang, Gu Ruobo, Xu Junmin, Yang Jia'an, Fan Chongxu, Yao Ge, Chen Ji-Sheng, Jiang Hui, Gao Bingmiao, Shi Qiong
Shenzhen Key Lab of Marine Genomics, Guangdong Provincial Key Lab of Molecular Breeding in Marine Economic Animals, BGI Academy of Marine Sciences, BGI Marine, BGI, Shenzhen, Guangdong, 518083, China.
Key Laboratory of Tropical Translational Medicine of Ministry of Education, Hainan Provincial Key Laboratory of Research and Development of Herbs, School of Pharmacy, Hainan Medical University, Haikou, Hainan, 570102, China.
Cell Discov. 2021 Feb 23;7(1):11. doi: 10.1038/s41421-021-00244-7.
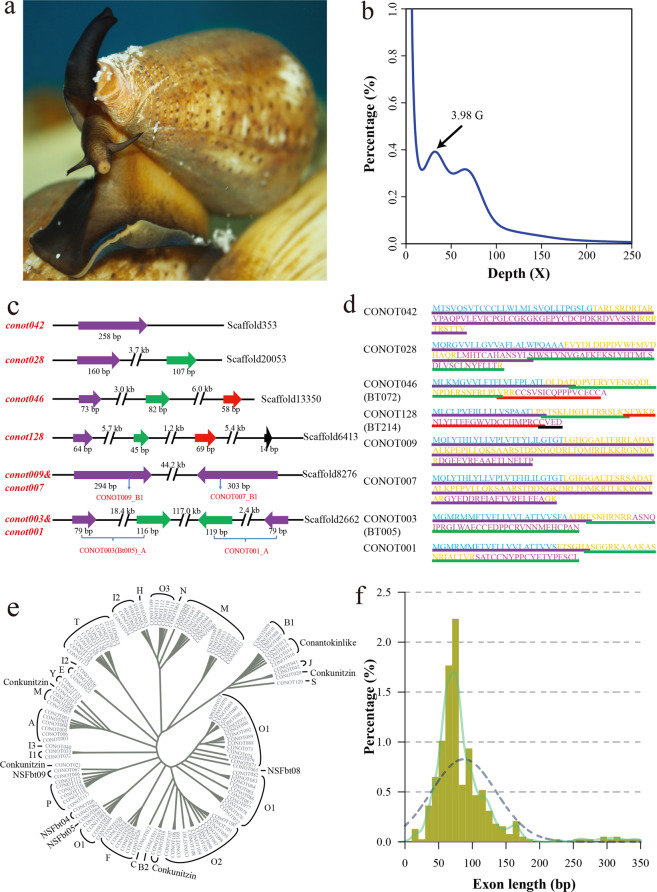
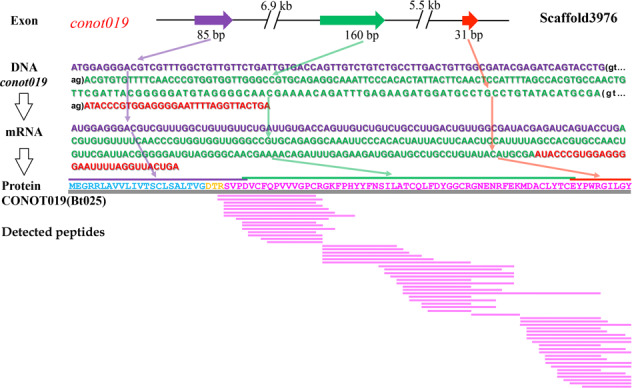
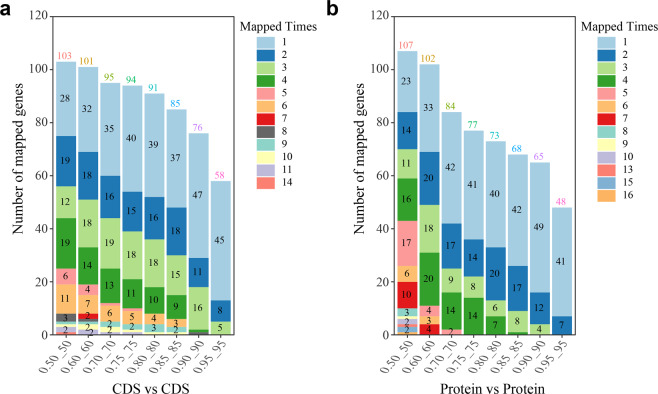
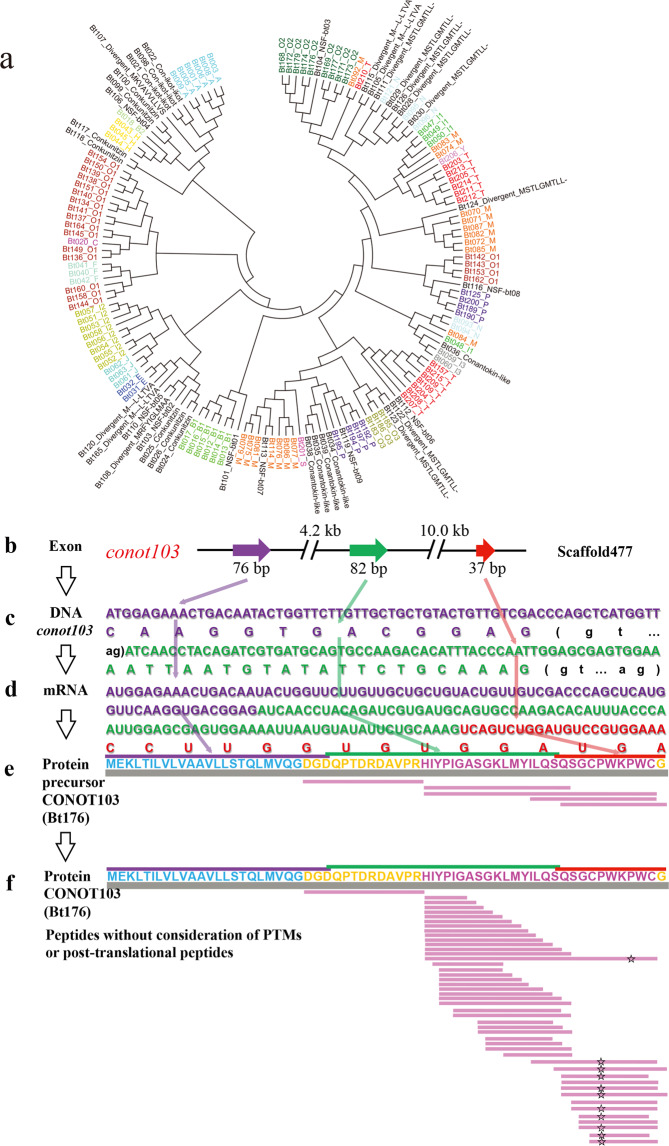
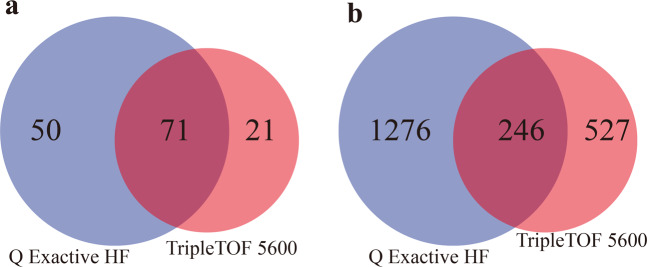
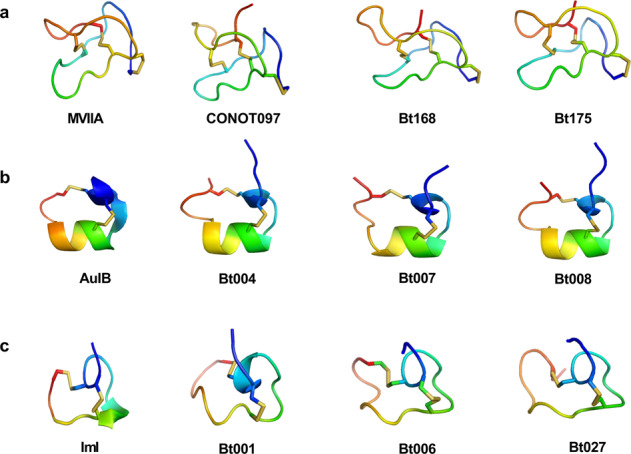
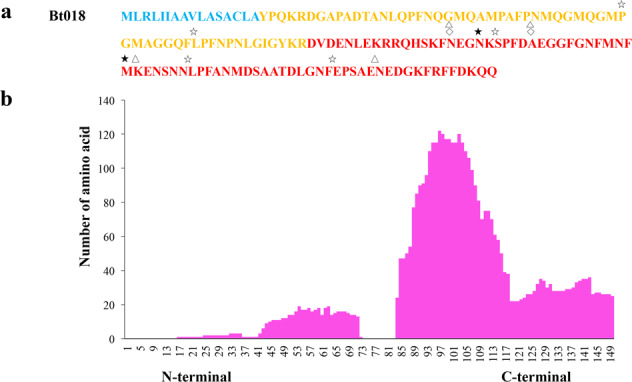
Although there are various Conus species with publicly available transcriptome and proteome data, no genome assembly has been reported yet. Here, using Chinese tubular cone snail (C. betulinus) as a representative, we sequenced and assembled the first Conus genome with original identification of 133 genome-widely distributed conopeptide genes. After integration of our genomics, transcriptomics, and peptidomics data in the same species, we established a primary genetic central dogma of diverse conopeptides, assuming a rough number ratio of ~1:1:1:10s for the total genes: transcripts: proteins: post-translationally modified peptides. This ratio may be special for this worm-hunting Conus species, due to the high diversity of various Conus genomes and the big number ranges of conopeptide genes, transcripts, and peptides in previous reports of diverse Conus species. Only a fraction (45.9%) of the identified conotopeptide genes from our achieved genome assembly are transcribed with transcriptomic evidence, and few genes individually correspond to multiple transcripts possibly due to intraspecies or mutation-based variances. Variable peptide processing at the proteomic level, generating a big diversity of venom conopeptides with alternative cleavage sites, post-translational modifications, and N-/C-terminal truncations, may explain how the 133 genes and ~123 transcripts can generate thousands of conopeptides in the venom of individual C. betulinus. We also predicted many conopeptides with high stereostructural similarities to the putative analgesic ω-MVIIA, addiction therapy AuIB and insecticide ImI, suggesting that our current genome assembly for C. betulinus is a valuable genetic resource for high-throughput prediction and development of potential pharmaceuticals.
尽管有多种芋螺物种拥有公开可用的转录组和蛋白质组数据,但尚未有基因组组装的报道。在此,我们以中国管芋螺(C. betulinus)为代表,对首个芋螺基因组进行了测序和组装,并首次鉴定出133个在全基因组广泛分布的芋螺毒素基因。在整合了同一物种的基因组学、转录组学和肽组学数据后,我们建立了一个关于多种芋螺毒素的初步遗传中心法则,假定总基因、转录本、蛋白质和翻译后修饰肽的数量比例大致为1:1:1:10。由于各种芋螺基因组的高度多样性以及之前报道的不同芋螺物种中芋螺毒素基因、转录本和肽的数量范围很大,这个比例可能对这种以蠕虫为食的芋螺物种来说是特殊的。在我们获得的基因组组装中,仅45.9%的已鉴定芋螺毒素基因有转录组证据表明其被转录,并且由于种内或基于突变的差异,很少有基因单独对应多个转录本。蛋白质组水平上可变的肽加工过程,产生了具有不同切割位点、翻译后修饰以及N-/C-末端截短的大量毒液芋螺毒素,这或许可以解释133个基因和约123个转录本如何在单个中国管芋螺的毒液中产生数千种芋螺毒素。我们还预测了许多与假定的镇痛剂ω-MVIIA、成瘾治疗药物AuIB和杀虫剂ImI具有高度立体结构相似性的芋螺毒素,这表明我们目前对中国管芋螺的基因组组装是用于高通量预测和开发潜在药物的宝贵遗传资源。