Institute of Molecular Biology, Academy of Sciences of Republic of Armenia, Yerevan, Armenia.
Division of Molecular and Cellular Biosciences, National Science Foundation, Alexandria, USA.
F1000Res. 2021 Jan 5;10:3. doi: 10.12688/f1000research.28175.1. eCollection 2021.
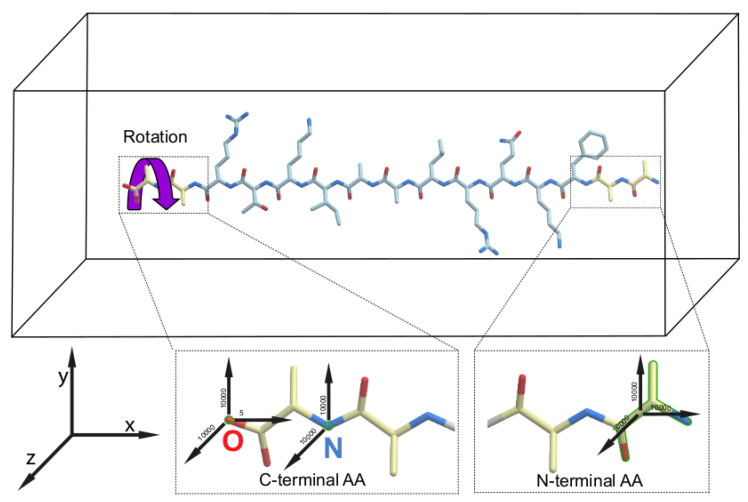
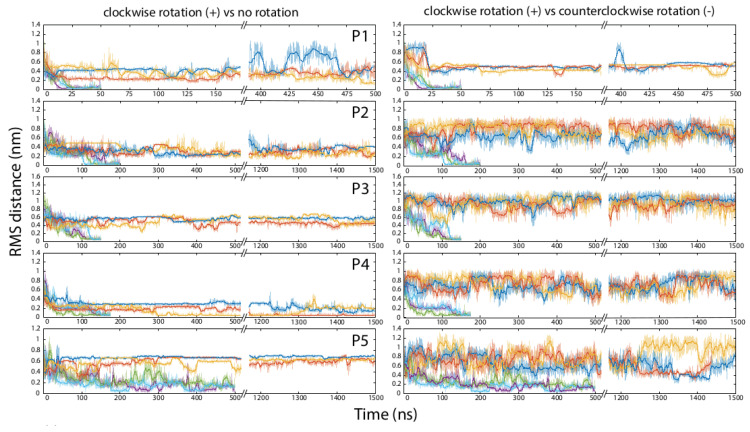
Proteins fold robustly and reproducibly , but many cannot fold in isolation from cellular components. Despite the remarkable progress that has been achieved by the artificial intelligence approaches in predicting the protein native conformations, the pathways that lead to such conformations, either or , remain largely unknown. The slow progress in recapitulating protein folding pathways may be an indication of the fundamental deficiencies in our understanding of folding as it occurs in nature. Here we consider the possibility that protein folding in living cells may not be driven solely by the decrease in Gibbs free energy and propose that protein folding should be modeled as an active energy-dependent process. The mechanism of action of such a protein folding machine might include direct manipulation of the peptide backbone. To show the feasibility of a protein folding machine, we conducted molecular dynamics simulations that were augmented by the application of mechanical force to rotate the C-terminal amino acid while simultaneously limiting the N-terminal amino acid movements. Remarkably, the addition of this simple manipulation of peptide backbones to the standard molecular dynamics simulation indeed facilitated the formation of native structures in five diverse alpha-helical peptides. Steric clashes that arise in the peptides due to the forced directional rotation resulted in the behavior of the peptide backbone no longer resembling a freely jointed chain. These simulations show the feasibility of a protein folding machine operating under the conditions when the movements of the polypeptide backbone are restricted by applying external forces and constraints. Further investigation is needed to see whether such an effect may play a role during co-translational protein folding and how it can be utilized to facilitate folding of proteins in artificial environments.
蛋白质能够稳健且可重复地折叠,但许多蛋白质无法在与细胞成分隔离的情况下折叠。尽管人工智能方法在预测蛋白质天然构象方面取得了显著进展,但导致这些构象的途径,无论是折叠途径还是未折叠途径,在很大程度上仍然未知。在重现蛋白质折叠途径方面进展缓慢,可能表明我们对自然发生的折叠的理解存在根本缺陷。在这里,我们考虑了这样一种可能性,即在活细胞中,蛋白质折叠可能不仅仅是由吉布斯自由能降低驱动的,并提出蛋白质折叠应该被建模为一个依赖能量的主动过程。这种蛋白质折叠机器的作用机制可能包括对肽主链的直接操作。为了展示蛋白质折叠机器的可行性,我们进行了分子动力学模拟,同时应用机械力旋转 C 末端氨基酸,同时限制 N 末端氨基酸的运动。值得注意的是,将这种简单的肽主链操作添加到标准分子动力学模拟中,确实有助于五种不同的α-螺旋肽形成天然结构。由于强制定向旋转而在肽中产生的空间位阻导致肽主链的行为不再类似于自由连接链。这些模拟表明,在施加外部力和约束限制多肽主链运动的条件下,蛋白质折叠机器是可行的。需要进一步研究以确定这种效应是否可能在共翻译蛋白质折叠过程中发挥作用,以及如何利用它来促进人工环境中蛋白质的折叠。