Strenic LLC, McLean, VA 22102, USA.
Division of Molecular and Cellular Biosciences, National Science Foundation, Alexandria, VA 22314, USA.
Int J Mol Sci. 2022 Jan 4;23(1):521. doi: 10.3390/ijms23010521.
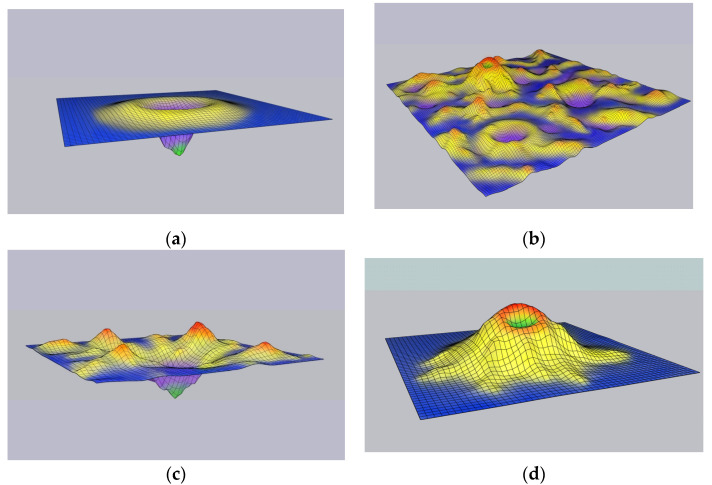
The prevailing current view of protein folding is the thermodynamic hypothesis, under which the native folded conformation of a protein corresponds to the global minimum of Gibbs free energy . We question this concept and show that the empirical evidence behind the thermodynamic hypothesis of folding is far from strong. Furthermore, physical theory-based approaches to the prediction of protein folds and their folding pathways so far have invariably failed except for some very small proteins, despite decades of intensive theory development and the enormous increase of computer power. The recent spectacular successes in protein structure prediction owe to evolutionary modeling of amino acid sequence substitutions enhanced by deep learning methods, but even these breakthroughs provide no information on the protein folding mechanisms and pathways. We discuss an alternative view of protein folding, under which the native state of most proteins does not occupy the global free energy minimum, but rather, a local minimum on a fluctuating free energy landscape. We further argue that Δ of folding is likely to be positive for the majority of proteins, which therefore fold into their native conformations only through interactions with the energy-dependent molecular machinery of living cells, in particular, the translation system and chaperones. Accordingly, protein folding should be modeled as it occurs in vivo, that is, as a non-equilibrium, active, energy-dependent process.
目前普遍认为蛋白质折叠遵循热力学假说,根据该假说,蛋白质的天然折叠构象对应于吉布斯自由能的全局最小值。我们对这一概念提出质疑,并指出折叠热力学假说背后的经验证据远不够充分。此外,基于物理理论的蛋白质折叠及其折叠途径的预测方法迄今无一成功,除了一些非常小的蛋白质,尽管经过几十年的理论发展和计算机能力的巨大提高。最近在蛋白质结构预测方面的惊人成功归功于通过深度学习方法增强的氨基酸序列取代的进化建模,但即使是这些突破也没有提供关于蛋白质折叠机制和途径的信息。我们讨论了一种蛋白质折叠的替代观点,根据该观点,大多数蛋白质的天然状态并不占据全局自由能最小值,而是在不断变化的自由能景观上的局部最小值。我们进一步认为,大多数蛋白质的折叠变化量Δ可能为正,因此,它们只有通过与活细胞中依赖能量的分子机制(特别是翻译系统和伴侣蛋白)相互作用,才能折叠成其天然构象。因此,蛋白质折叠应该在体内发生时进行建模,也就是说,作为一个非平衡、主动、依赖能量的过程。