Felber Veronika Barbara, Valentin Manuel Amando, Wester Hans-Jürgen
Technical University of Munich, Chair of Pharmaceutical Radiochemistry, Walther-Meißner-Str. 3, 85748, Garching, Germany.
EJNMMI Radiopharm Chem. 2021 Feb 26;6(1):10. doi: 10.1186/s41181-021-00124-1.
To investigate whether modifications of prostate-specific membrane antigen (PSMA)-targeted radiolabeled urea-based inhibitors could reduce salivary gland uptake and thus improve tumor-to-salivary gland ratios, several analogs of a high affinity PSMA ligand were synthesized and evaluated in in vitro and in vivo studies.
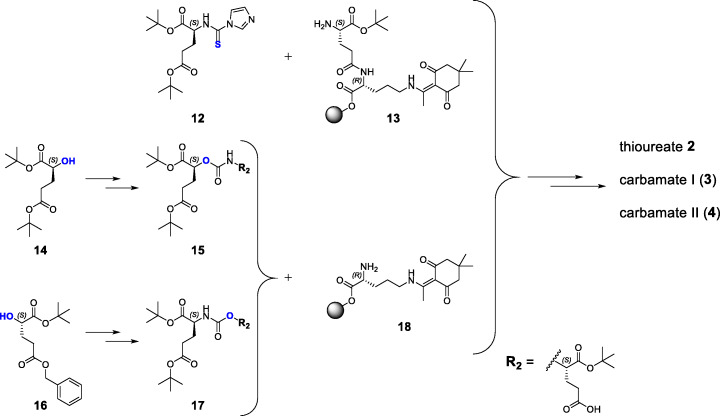
Binding motifs were synthesized 'on-resin' or, when not practicable, in solution. Peptide chain elongations were performed according to optimized standard protocols via solid-phase peptide synthesis. In vitro experiments were performed using PSMA LNCaP cells. In vivo studies as well as μSPECT/CT scans were conducted with male LNCaP tumor xenograft-bearing CB17-SCID mice.
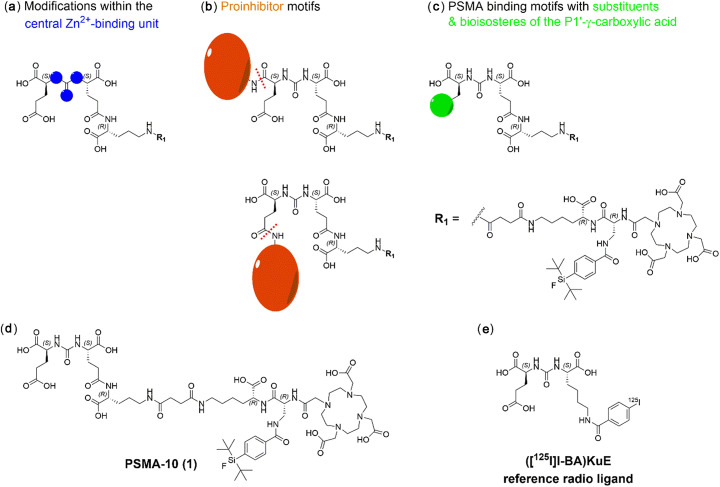
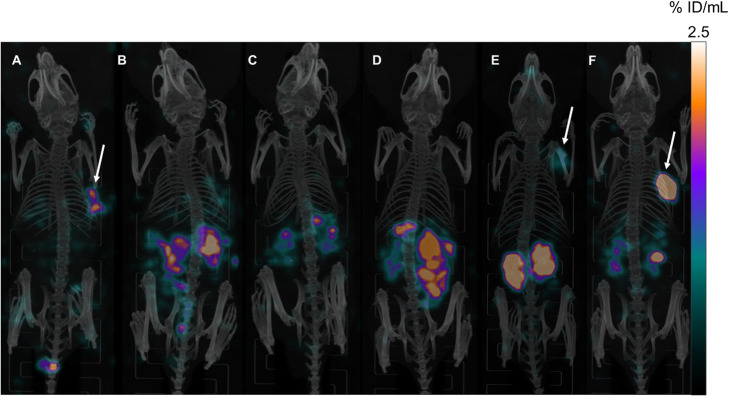
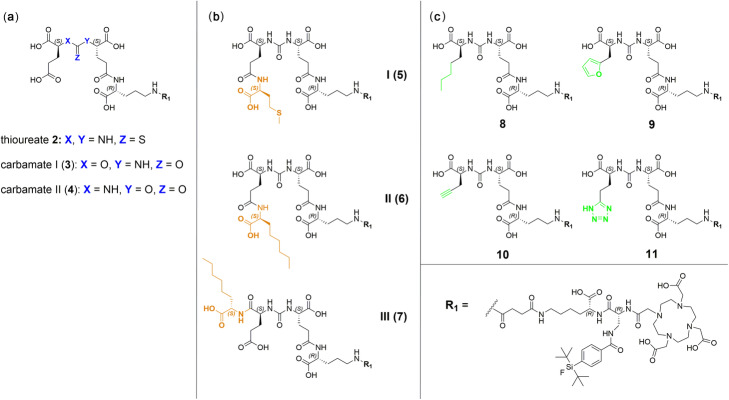
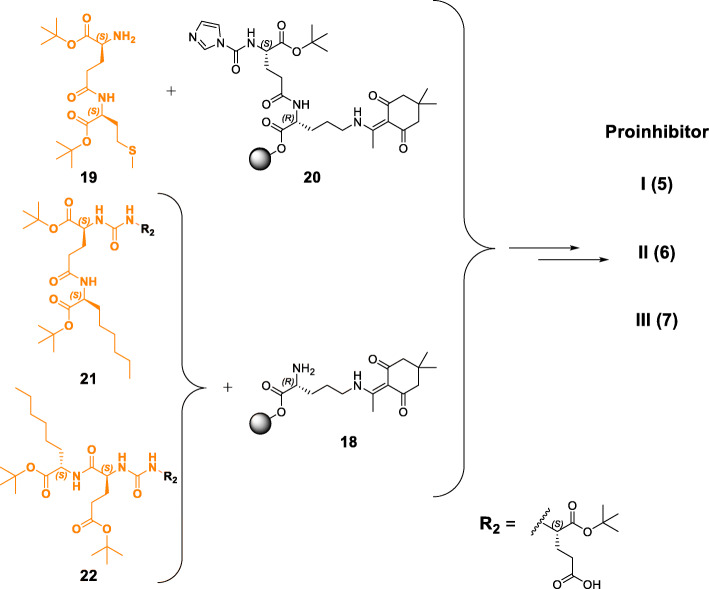
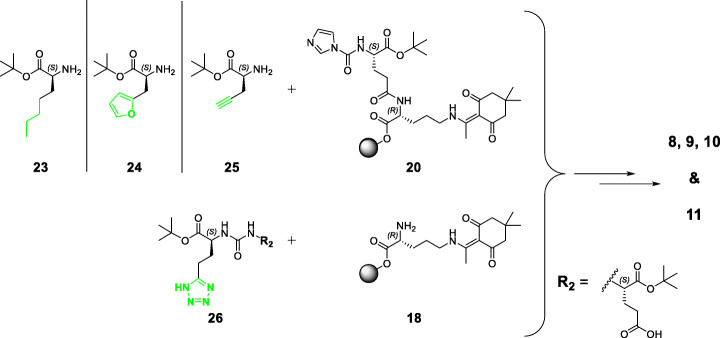
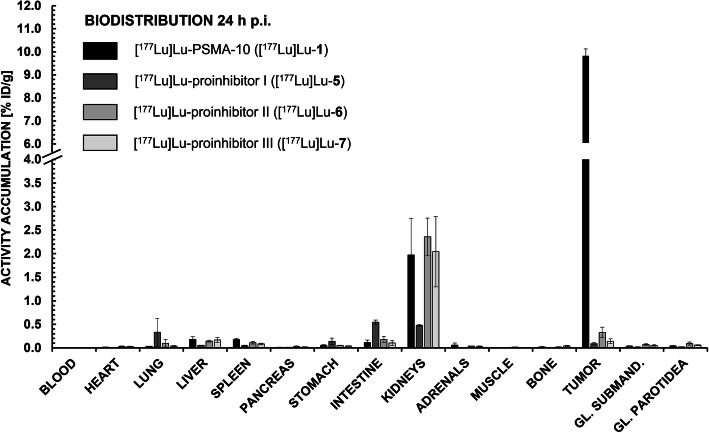
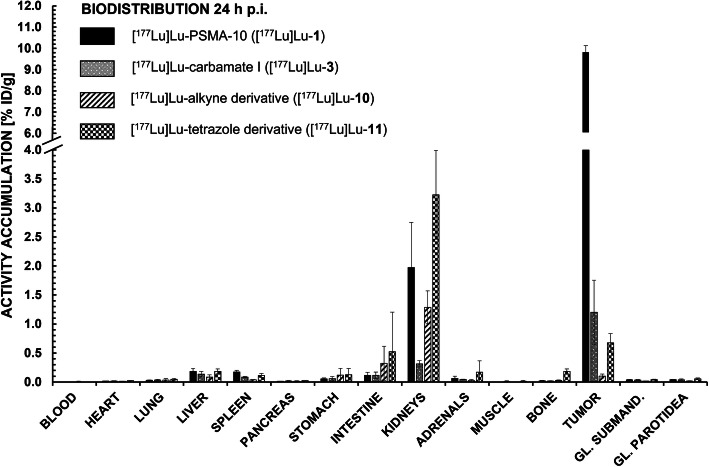
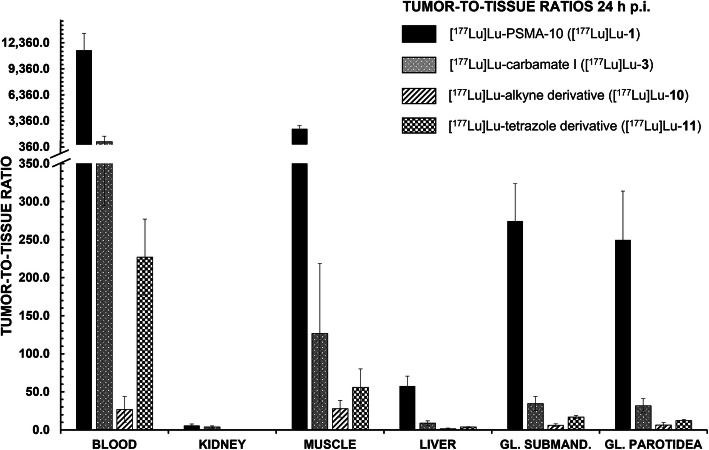
PSMA ligands with A) modifications within the central Zn-binding unit, B) proinhibitor motifs and C) substituents & bioisosteres of the P1'-γ-carboxylic acid were synthesized and evaluated. Modifications within the central Zn-binding unit of PSMA-10 (Glu-urea-Glu) provided three compounds. Thereof, only Lu-carbamate I (Lu-3) exhibited high affinity (IC = 7.1 ± 0.7 nM), but low tumor uptake (5.31 ± 0.94% ID/g, 1 h p.i. and 1.20 ± 0.55% ID/g, 24 h p.i.). All proinhibitor motif-based ligands (three in total) exhibited low binding affinities (> 1 μM), no notable internalization and very low tumor uptake (< 0.50% ID/g). In addition, four compounds with P1'-ɣ-carboxylate substituents were developed and evaluated. Thereof, only tetrazole derivative Lu-11 revealed high affinity (IC = 16.4 ± 3.8 nM), but also this inhibitor showed low tumor uptake (3.40 ± 0.63% ID/g, 1 h p.i. and 0.68 ± 0.16% ID/g, 24 h p.i.). Salivary gland uptake in mice remained at an equally low level for all compounds (between 0.02 ± 0.00% ID/g and 0.09 ± 0.03% ID/g), wherefore apparent tumor-to-submandibular gland and tumor-to-parotid gland ratios for the modified peptides were distinctly lower (factor 8-45) than for [Lu]Lu-PSMA-10 at 24 h p.i.
The investigated compounds could not compete with the in vivo characteristics of the EuE-based PSMA inhibitor [Lu]Lu-PSMA-10. Although two derivatives (3 and 11) were found to exhibit high affinities towards LNCaP cells, tumor uptake at 24 h p.i. was considerably low, while uptake in salivary glands remained unaffected. Optimization of the established animal model should be envisaged to enable a clear identification of PSMA-targeting radioligands with improved tumor-to-salivary gland ratios in future studies.
为研究对前列腺特异性膜抗原(PSMA)靶向放射性标记的脲基抑制剂进行修饰是否能够降低唾液腺摄取,从而提高肿瘤与唾液腺的比值,合成了一种高亲和力PSMA配体的几种类似物,并进行了体外和体内研究。
结合基序在树脂上合成,或在不可行时在溶液中合成。通过固相肽合成,根据优化的标准方案进行肽链延伸。使用PSMA LNCaP细胞进行体外实验。对雄性LNCaP肿瘤异种移植CB17-SCID小鼠进行体内研究以及μSPECT/CT扫描。
合成并评估了具有以下特点的PSMA配体:A)中心锌结合单元内的修饰;B)前抑制剂基序;C)P1'-γ-羧酸的取代基和生物电子等排体。PSMA-10(Glu-脲-Glu)中心锌结合单元内的修饰产生了三种化合物。其中,只有氨基甲酸镥I(Lu-3)表现出高亲和力(IC = 7.1±0.7 nM),但肿瘤摄取较低(注射后1小时为5.31±0.94% ID/g,注射后24小时为1.20±0.55% ID/g)。所有基于前抑制剂基序的配体(共三种)均表现出低结合亲和力(>1 μM),无明显内化且肿瘤摄取极低(<0.50% ID/g)。此外,开发并评估了四种具有P1'-γ-羧酸盐取代基的化合物。其中,只有四唑衍生物Lu-11表现出高亲和力(IC = 16.4±3.8 nM),但该抑制剂的肿瘤摄取也较低(注射后1小时为3.40±0.63% ID/g,注射后24小时为0.68±0.16% ID/g)。所有化合物在小鼠唾液腺中的摄取均维持在同样低的水平(0.02±0.00% ID/g至0.09±0.03% ID/g之间),因此修饰后肽段的肿瘤与下颌下腺以及肿瘤与腮腺的表观比值在注射后24小时明显低于[Lu]Lu-PSMA-10(8至45倍)。
所研究的化合物无法与基于EuE的PSMA抑制剂[Lu]Lu-PSMA-10的体内特性相竞争。尽管发现两种衍生物(3和11)对LNCaP细胞具有高亲和力,但注射后24小时的肿瘤摄取相当低,而唾液腺摄取未受影响。应设想对已建立的动物模型进行优化,以便在未来研究中能够明确鉴定出具有改善的肿瘤与唾液腺比值的PSMA靶向放射性配体。