Yepes Manuel
Department of Neurology, Emory University School of Medicine; Department of Neurology, Veterans Affairs Medical Center; Division of Neuropharmacology and Neurologic Diseases, Yerkes National Primate Research Center, Atlanta, GA, USA.
Neural Regen Res. 2021 Oct;16(10):1973-1977. doi: 10.4103/1673-5374.308076.
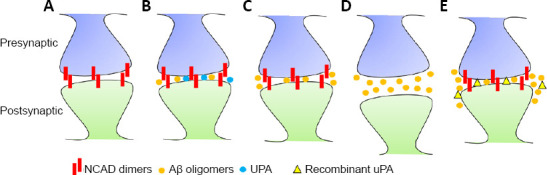
Dementia is a clinical syndrome that affects approximately 47 million people worldwide and is characterized by progressive and irreversible decline of cognitive, behavioral and sesorimotor functions. Alzheimer's disease (AD) accounts for approximately 60-80% of all cases of dementia, and neuropathologically is characterized by extracellular deposits of insoluble amyloid-β (Aβ) and intracellular aggregates of hyperphosphorylated tau. Significantly, although for a long time it was believed that the extracellular accumulation of Aβ was the culprit of the symptoms observed in these patients, more recent studies have shown that cognitive decline in people suffering this disease is associated with soluble Aβ-induced synaptic dysfunction instead of the formation of insoluble Aβ-containing extracellular plaques. These observations are translationally relevant because soluble Aβ-induced synaptic dysfunction is an early event in AD that precedes neuronal death, and thus is amenable to therapeutic interventions to prevent cognitive decline before the progression to irreversible brain damage. The plasminogen activating (PA) system is an enzymatic cascade that triggers the degradation of fibrin by catalyzing the conversion of plasminogen into plasmin via two serine proteinases: tissue-type plasminogen activator (tPA) and urokinase-type plasminogen activator (uPA). Experimental evidence reported over the last three decades has shown that tPA and uPA play a role in the pathogenesis of AD. However, these studies have focused on the ability of these plasminogen activators to trigger plasmin-induced cleavage of insoluble Aβ-containing extracellular plaques. In contrast, recent evidence indicates that activity-dependent release of uPA from the presynaptic terminal of cerebral cortical neurons protects the synapse from the deleterious effects of soluble Aβ via a mechanism that does not require plasmin generation or the cleavage of Aβ fibrils. Below we discuss the role of the PA system in the pathogenesis of AD and the translational relevance of data published to this date.
痴呆是一种临床综合征,全球约有4700万人受其影响,其特征是认知、行为和感觉运动功能进行性且不可逆地衰退。阿尔茨海默病(AD)约占所有痴呆病例的60%-80%,其神经病理学特征是细胞外不溶性淀粉样β蛋白(Aβ)沉积和细胞内过度磷酸化tau蛋白聚集。值得注意的是,尽管长期以来人们认为Aβ的细胞外积累是这些患者出现症状的罪魁祸首,但最近的研究表明,患这种疾病的人的认知衰退与可溶性Aβ诱导的突触功能障碍有关,而不是与含不溶性Aβ的细胞外斑块形成有关。这些观察结果具有转化相关性,因为可溶性Aβ诱导的突触功能障碍是AD中的早期事件,发生在神经元死亡之前,因此适合在进展到不可逆脑损伤之前进行预防认知衰退的治疗干预。纤溶酶原激活(PA)系统是一种酶促级联反应,通过两种丝氨酸蛋白酶:组织型纤溶酶原激活剂(tPA)和尿激酶型纤溶酶原激活剂(uPA)催化纤溶酶原转化为纤溶酶,从而触发纤维蛋白的降解。过去三十年报道的实验证据表明,tPA和uPA在AD的发病机制中起作用。然而,这些研究集中在这些纤溶酶原激活剂触发纤溶酶诱导的含不溶性Aβ的细胞外斑块裂解的能力上。相比之下,最近的证据表明,大脑皮质神经元突触前末端uPA的活性依赖性释放通过一种不需要纤溶酶生成或Aβ纤维裂解的机制保护突触免受可溶性Aβ的有害影响。下面我们讨论PA系统在AD发病机制中的作用以及迄今为止发表的数据的转化相关性。