Graduate School, Inner Mongolia Medical University, Hohhot, 010059, Inner Mongolia, China.
Department of Parasitology, Inner Mongolia Medical University, Hohhot, 010110, Inner Mongolia, China.
Parasit Vectors. 2021 Mar 1;14(1):131. doi: 10.1186/s13071-021-04625-5.
Ticks (Arthropoda, Ixodida), after mosquitoes, are the second most prevalent vector of infectious diseases. They are responsible for spreading a multitude of pathogens and threatening the health and welfare of animals and human beings. However, given the history of tick-borne pathogen infections in the Inner Mongolia Autonomous Region of China, surprisingly, neither the genetic diversity nor the spatial distribution of haplotypes within ticks has been studied.
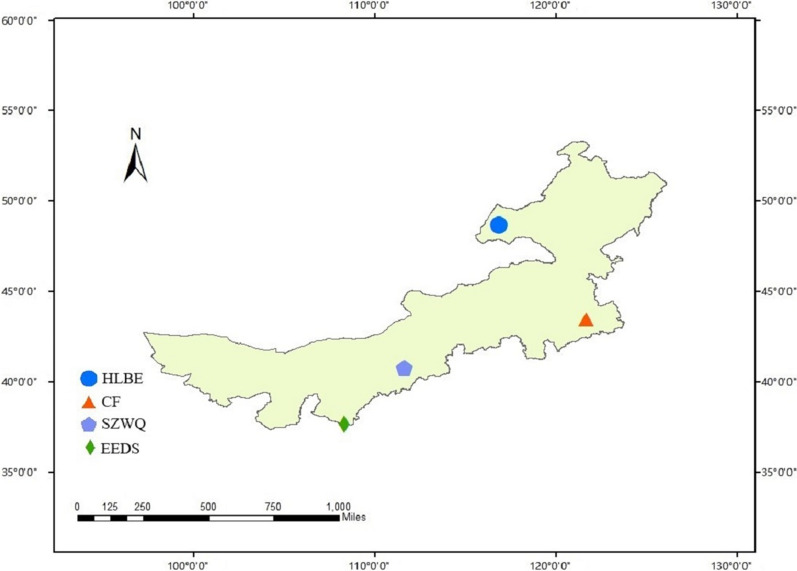
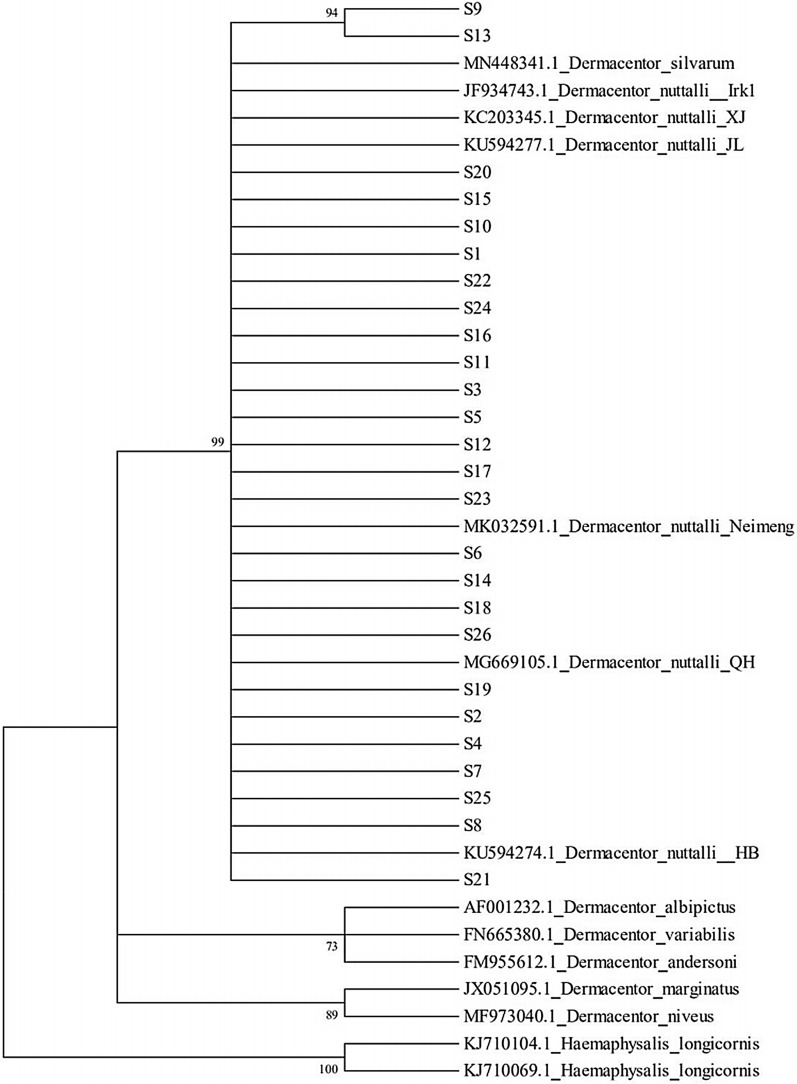
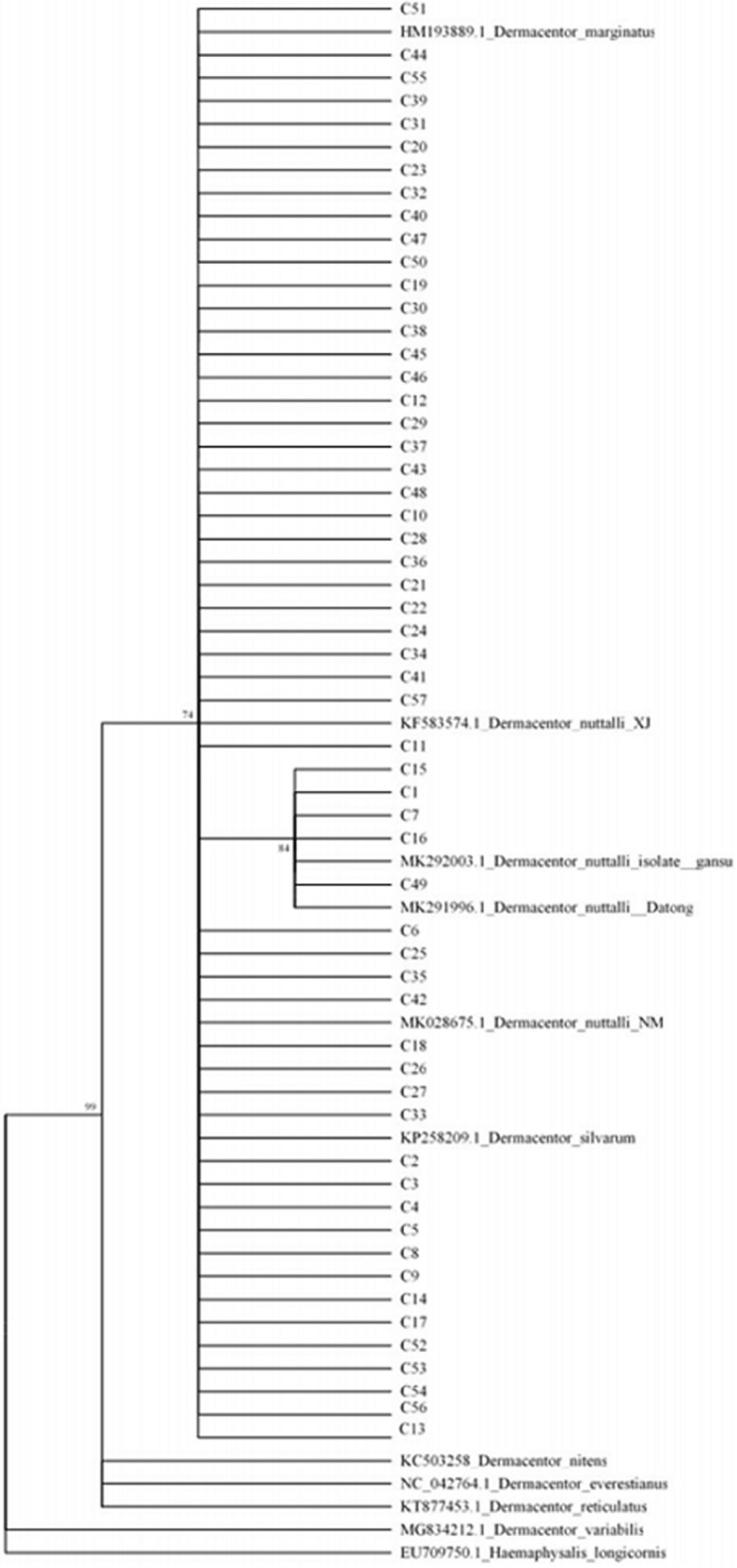
We characterized the haplotype distribution of Dermacentor nuttalli in four main pastoral areas of the Inner Mongolia Autonomous Region, by sampling 109 individuals (recovered from sheep) in April-August 2019. The 16S rRNA gene, cytochrome c oxidase subunit I (COI), and the internal transcribed spacer 2 region (ITS2) were amplified and sequenced from extracted DNA.
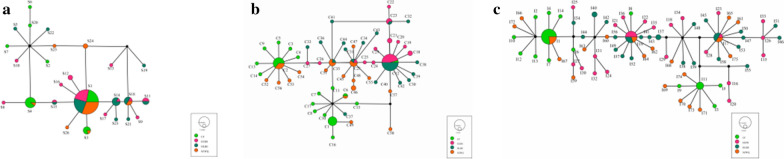
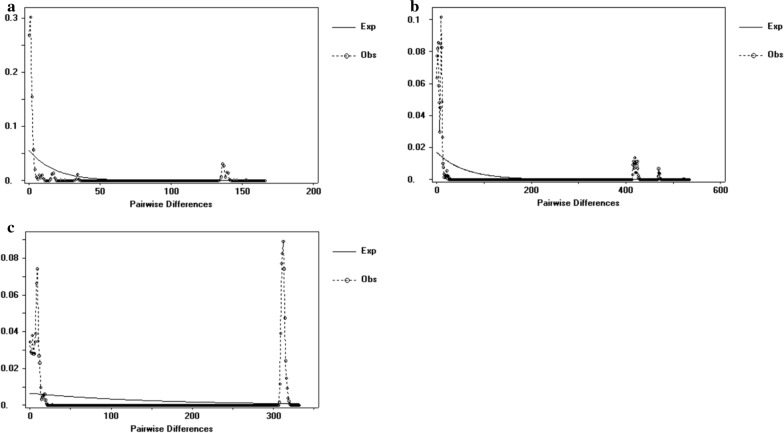
Twenty-six haplotypes were identified using 16S rRNA sequences, 57 haplotypes were identified with COI sequences, and 75 haplotypes were identified with ITS2 sequences. Among the three genes, total haplotype diversity was greater than 0.7, while total nucleotide diversity was greater than 0.06. Neutrality tests revealed a significantly negative Tajima's D result, while Fu's Fs was not significantly positive. Fixation index values (F) indicated that the degree of genetic differentiation among some sampled populations was small, while for others it was moderate. Analysis of molecular variance (AMOVA) revealed that the variation within populations was greater than that among populations. The mismatch analysis of D. nuttalli exhibited double peaks.
The genetic diversity of D. nuttalli populations in our region can likely adapt to different geographical environments, thereby leading to genetic diversity, and creating genetic differentiation among different populations. However, genetic differentiation is cryptic and does not form a pedigree geographical structure.
蜱虫(节肢动物门,硬蜱目)是继蚊子之后第二大常见的传染病媒介。它们传播了大量的病原体,威胁着动物和人类的健康和福利。然而,鉴于中国内蒙古自治区蜱传病原体感染的历史,令人惊讶的是,蜱虫内部的遗传多样性和单倍型空间分布都没有得到研究。
我们通过在 2019 年 4 月至 8 月从绵羊身上采集的 109 个样本,对内蒙古自治区四个主要牧区的钝缘蜱进行了单倍型分布特征分析。从提取的 DNA 中扩增和测序了 16S rRNA 基因、细胞色素 c 氧化酶亚基 I(COI)和内部转录间隔区 2 区(ITS2)。
使用 16S rRNA 序列鉴定出 26 个单倍型,使用 COI 序列鉴定出 57 个单倍型,使用 ITS2 序列鉴定出 75 个单倍型。在这三个基因中,总单倍型多样性大于 0.7,而总核苷酸多样性大于 0.06。中性检验显示 Tajima's D 结果显著为负,而 Fu's Fs 不显著为正。固定指数(F)值表明,一些采样种群的遗传分化程度较小,而其他种群的遗传分化程度中等。分析分子方差(AMOVA)显示,种群内的变异大于种群间的变异。钝缘蜱的错配分析呈双峰状。
我们研究区域内钝缘蜱种群的遗传多样性可能能够适应不同的地理环境,从而导致遗传多样性,并在不同种群之间产生遗传分化。然而,遗传分化是隐匿的,没有形成谱系地理结构。