Department of Anesthesiology, Perioperative and Pain Medicine, Stanford University School of Medicine, Stanford, CA, USA.
Department of Obstetrics and Gynecology, Wayne State University School of Medicine, Detroit, MI, USA.
J Matern Fetal Neonatal Med. 2022 Dec;35(25):5621-5628. doi: 10.1080/14767058.2021.1888915. Epub 2021 Mar 2.
Early identification of pregnant women at risk for preeclampsia (PE) is important, as it will enable targeted interventions ahead of clinical manifestations. The quantitative analyses of plasma proteins feature prominently among molecular approaches used for risk prediction. However, derivation of protein signatures of sufficient predictive power has been challenging. The recent availability of platforms simultaneously assessing over 1000 plasma proteins offers broad examinations of the plasma proteome, which may enable the extraction of proteomic signatures with improved prognostic performance in prenatal care.
The primary aim of this study was to examine the generalizability of proteomic signatures predictive of PE in two cohorts of pregnant women whose plasma proteome was interrogated with the same highly multiplexed platform. Establishing generalizability, or lack thereof, is critical to devise strategies facilitating the development of clinically useful predictive tests. A second aim was to examine the generalizability of protein signatures predictive of gestational age (GA) in uncomplicated pregnancies in the same cohorts to contrast physiological and pathological pregnancy outcomes.
Serial blood samples were collected during the first, second, and third trimesters in 18 women who developed PE and 18 women with uncomplicated pregnancies (Stanford cohort). The second cohort (Detroit), used for comparative analysis, consisted of 76 women with PE and 90 women with uncomplicated pregnancies. Multivariate analyses were applied to infer predictive and cohort-specific proteomic models, which were then tested in the alternate cohort. Gene ontology (GO) analysis was performed to identify biological processes that were over-represented among top-ranked proteins associated with PE.
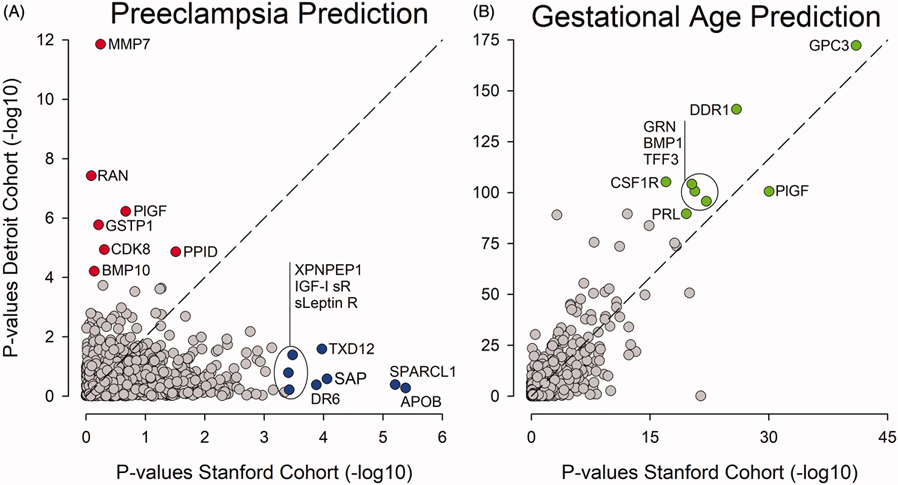
The model derived in the Stanford cohort was highly significant ( = 3.9E-15) and predictive (AUC = 0.96), but failed validation in the Detroit cohort ( = 9.7E-01, AUC = 0.50). Similarly, the model derived in the Detroit cohort was highly significant ( = 1.0E-21, AUC = 0.73), but failed validation in the Stanford cohort ( = 7.3E-02, AUC = 0.60). By contrast, proteomic models predicting GA were readily validated across the Stanford ( = 1.1E-454, = 0.92) and Detroit cohorts ( = 1.1.E-92, = 0.92) indicating that the proteomic assay performed well enough to infer a generalizable model across studied cohorts, which makes it less likely that technical aspects of the assay, including batch effects, accounted for observed differences.
Results point to a broader issue relevant for proteomic and other omic discovery studies in patient cohorts suffering from a clinical syndrome, such as PE, driven by heterogeneous pathophysiologies. While novel technologies including highly multiplex proteomic arrays and adapted computational algorithms allow for novel discoveries for a particular study cohort, they may not readily generalize across cohorts. A likely reason is that the prevalence of pathophysiologic processes leading up to the "same" clinical syndrome can be distributed differently in different and smaller-sized cohorts. Signatures derived in individual cohorts may simply capture different facets of the spectrum of pathophysiologic processes driving a syndrome. Our findings have important implications for the design of omic studies of a syndrome like PE. They highlight the need for performing such studies in diverse and well-phenotyped patient populations that are large enough to characterize subsets of patients with shared pathophysiologies to then derive subset-specific signatures of sufficient predictive power.
早期识别有先兆子痫 (PE) 风险的孕妇非常重要,因为这将使我们能够在临床表现出现之前进行有针对性的干预。定量分析血浆蛋白是用于风险预测的分子方法中的重要方法。然而,具有足够预测能力的蛋白质特征的推导一直具有挑战性。最近出现的同时评估 1000 多种血浆蛋白的平台提供了对血浆蛋白质组的广泛检查,这可能使我们能够提取具有改善产前护理预后性能的蛋白质特征。
本研究的主要目的是检查在两个孕妇队列中预测 PE 的蛋白质特征的可推广性,这些队列的血浆蛋白质组使用相同的高度多重化平台进行了检测。确定缺乏可推广性对于制定促进临床有用预测测试发展的策略至关重要。第二个目的是检查在同一队列中用于比较分析的无并发症妊娠中预测 GA 的蛋白质特征的可推广性,以对比生理和病理妊娠结果。
在斯坦福队列中,18 名患有 PE 的孕妇和 18 名无并发症妊娠的孕妇在第一、第二和第三个三个月采集了连续的血液样本。第二个队列(底特律)由 76 名患有 PE 的孕妇和 90 名无并发症妊娠的孕妇组成,用于比较分析。应用多元分析推断预测和队列特异性蛋白质模型,然后在替代队列中进行测试。进行基因本体论 (GO) 分析以确定与 PE 相关的排名最高的蛋白质中过度表达的生物学过程。
斯坦福队列中得出的模型具有高度显著性( = 3.9E-15)和预测性(AUC = 0.96),但在底特律队列中验证失败( = 9.7E-01,AUC = 0.50)。类似地,底特律队列中得出的模型具有高度显著性( = 1.0E-21,AUC = 0.73),但在斯坦福队列中验证失败( = 7.3E-02,AUC = 0.60)。相比之下,预测 GA 的蛋白质模型在斯坦福( = 1.1E-454, = 0.92)和底特律队列( = 1.1.E-92, = 0.92)中都很容易验证,这表明蛋白质分析的性能足以在研究队列中推断出可推广的模型,这使得分析技术方面(包括批次效应)不太可能导致观察到的差异。
结果表明,对于由异质病理生理学驱动的临床综合征(如 PE)的患者队列中的蛋白质组学和其他组学发现研究存在更广泛的问题。虽然包括高度多重蛋白质阵列和改编的计算算法在内的新技术允许为特定研究队列进行新的发现,但它们可能不容易在队列之间推广。一个可能的原因是,导致“相同”临床综合征的病理生理过程的流行程度在不同和较小的队列中可能分布不同。在各个队列中得出的特征可能只是捕获了驱动综合征的病理生理过程谱的不同方面。我们的研究结果对 PE 等综合征的组学研究设计具有重要意义。它们强调了在具有足够代表性的患者人群中进行此类研究的必要性,这些人群足够大,可以对具有共同病理生理学的患者亚群进行特征描述,然后得出具有足够预测能力的亚群特异性特征。