Ko Sung Hee, Mokhtari Elham Bayat, Mudvari Prakriti, Stein Sydney, Stringham Christopher D, Wagner Danielle, Ramelli Sabrina, Ramos-Benitez Marcos J, Strich Jeffrey R, Davey Richard T, Zhou Tongqing, Misasi John, Kwong Peter D, Chertow Daniel S, Sullivan Nancy J, Boritz Eli A
Vaccine Research Center, National Institute of Allergy and Infectious Diseases, National Institutes of Health, Bethesda, MD 20892, USA.
Emerging Pathogens Section, Critical Care Medicine Department, National Institutes of Health Clinical Center, Bethesda, MD 20892, USA.
bioRxiv. 2021 Feb 22:2021.02.21.432184. doi: 10.1101/2021.02.21.432184.
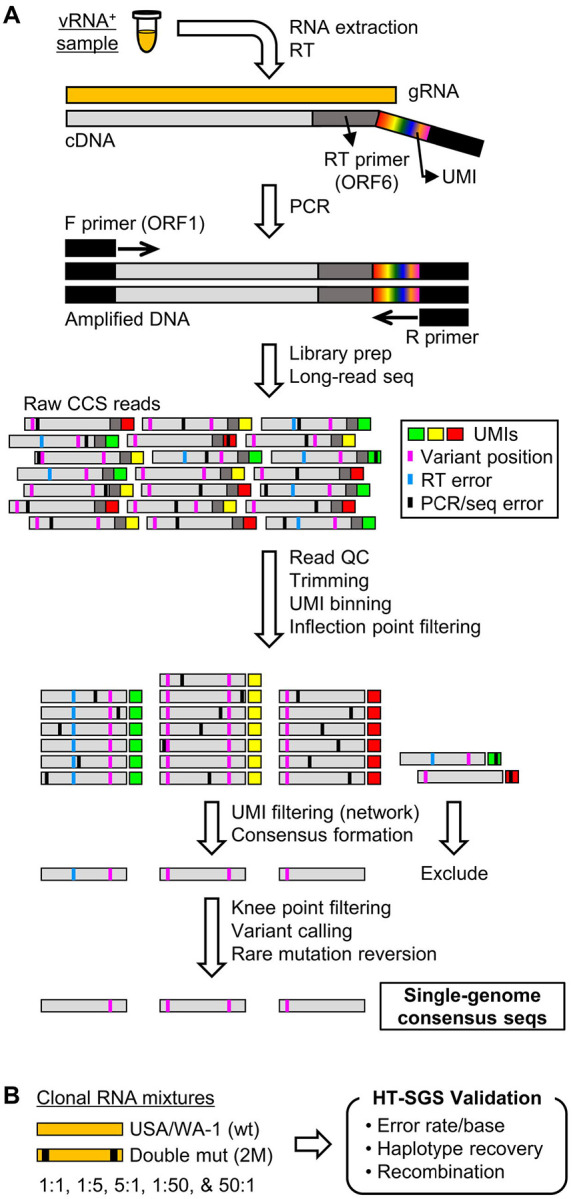
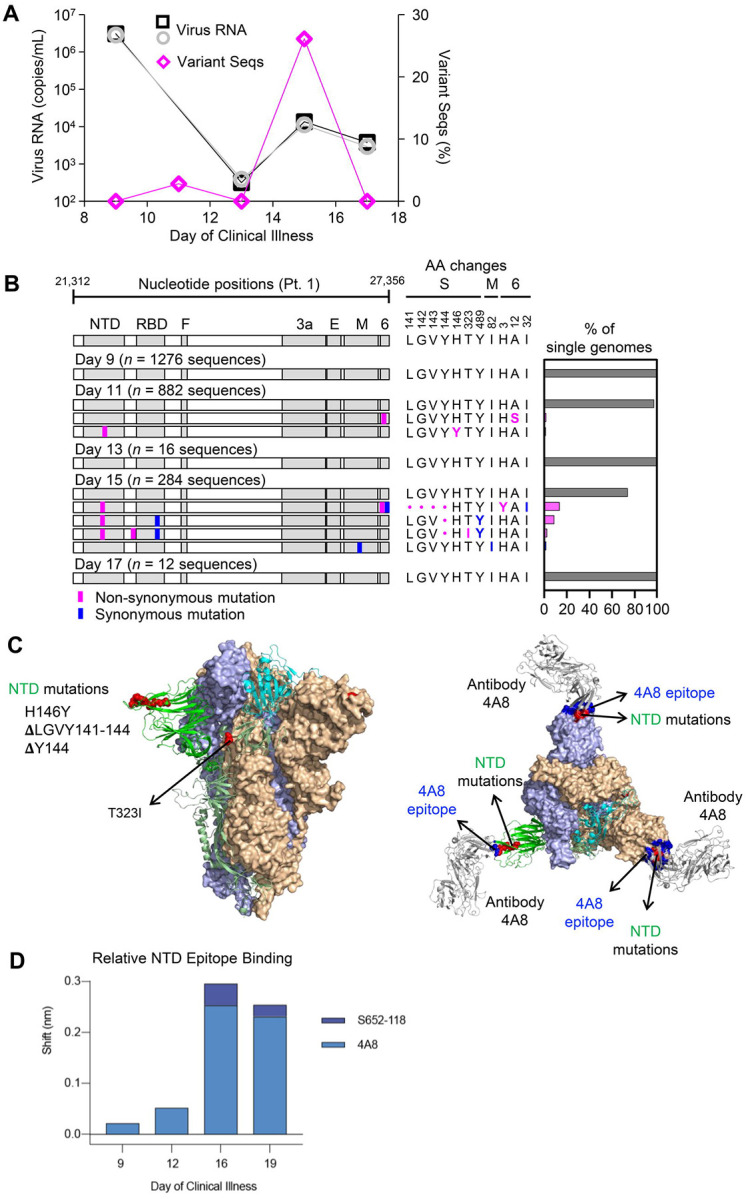
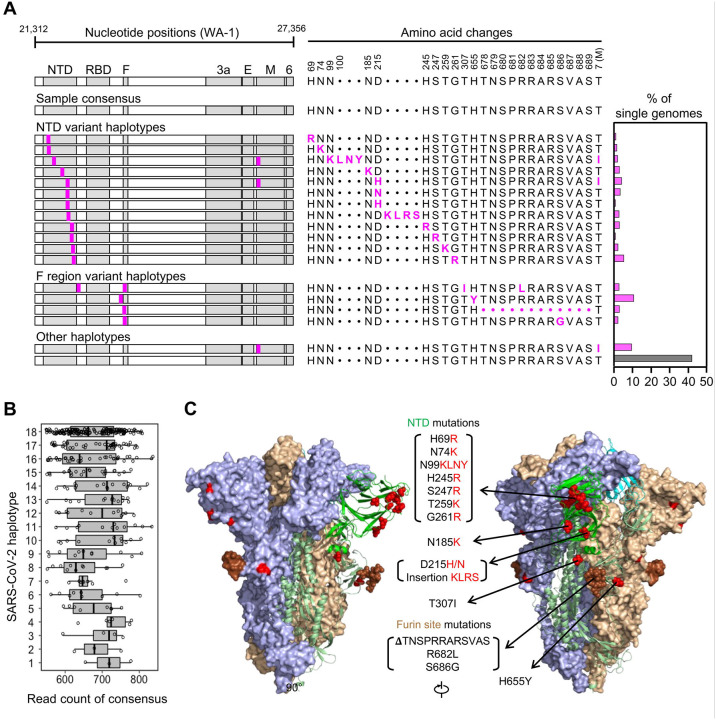
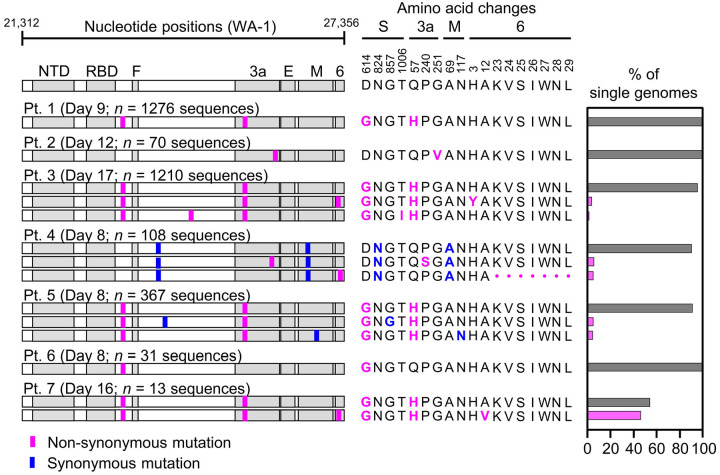
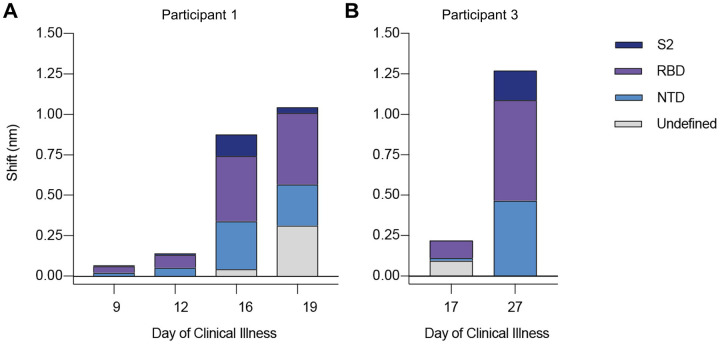
Tracking evolution of the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) within infected individuals will help elucidate coronavirus disease 2019 (COVID-19) pathogenesis and inform use of antiviral interventions. In this study, we developed an approach for sequencing the region encoding the SARS-CoV-2 virion surface proteins from large numbers of individual virus RNA genomes per sample. We applied this approach to the WA-1 reference clinical isolate of SARS-CoV-2 passaged and to upper respiratory samples from 7 study participants with COVID-19. SARS-CoV-2 genomes from cell culture were diverse, including 18 haplotypes with non-synonymous mutations clustered in the spike NH -terminal domain (NTD) and furin cleavage site regions. By contrast, cross-sectional analysis of samples from participants with COVID-19 showed fewer virus variants, without structural clustering of mutations. However, longitudinal analysis in one individual revealed 4 virus haplotypes bearing 3 independent mutations in a spike NTD epitope targeted by autologous antibodies. These mutations arose coincident with a 6.2-fold rise in serum binding to spike and a transient increase in virus burden. We conclude that SARS-CoV-2 exhibits a capacity for rapid genetic adaptation that becomes detectable with the onset of humoral immunity, with the potential to contribute to delayed virologic clearance in the acute setting.
Mutant sequences of severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2) arising during any individual case of coronavirus disease 2019 (COVID-19) could theoretically enable the virus to evade immune responses or antiviral therapies that target the predominant infecting virus sequence. However, commonly used sequencing technologies are not optimally designed to detect variant virus sequences within each sample. To address this issue, we developed novel technology for sequencing large numbers of individual SARS-CoV-2 genomic RNA molecules across the region encoding the virus surface proteins. This technology revealed extensive genetic diversity in cultured viruses from a clinical isolate of SARS-CoV-2, but lower diversity in samples from 7 individuals with COVID-19. Importantly, concurrent analysis of paired serum samples in selected individuals revealed relatively low levels of antibody binding to the SARS-CoV-2 spike protein at the time of initial sequencing. With increased serum binding to spike protein, we detected multiple SARS-CoV-2 variants bearing independent mutations in a single epitope, as well as a transient increase in virus burden. These findings suggest that SARS-CoV-2 replication creates sufficient virus genetic diversity to allow immune-mediated selection of variants within the time frame of acute COVID-19. Large-scale studies of SARS-CoV-2 variation and specific immune responses will help define the contributions of intra-individual SARS-CoV-2 evolution to COVID-19 clinical outcomes and antiviral drug susceptibility.
追踪严重急性呼吸综合征冠状病毒2(SARS-CoV-2)在受感染个体内的进化,将有助于阐明2019冠状病毒病(COVID-19)的发病机制,并为抗病毒干预措施的使用提供依据。在本研究中,我们开发了一种方法,用于对每个样本中大量个体病毒RNA基因组编码SARS-CoV-2病毒粒子表面蛋白的区域进行测序。我们将这种方法应用于传代的SARS-CoV-2 WA-1参考临床分离株,以及来自7名COVID-19研究参与者的上呼吸道样本。细胞培养中的SARS-CoV-2基因组具有多样性,包括18种单倍型,非同义突变聚集在刺突蛋白N端结构域(NTD)和弗林蛋白酶切割位点区域。相比之下,对COVID-19参与者样本的横断面分析显示病毒变体较少,且突变没有结构聚集。然而,对一名个体的纵向分析发现了4种病毒单倍型,在一种被自身抗体靶向的刺突NTD表位中有3个独立突变。这些突变与血清与刺突蛋白结合增加6.2倍以及病毒载量短暂增加同时出现。我们得出结论,SARS-CoV-2具有快速基因适应能力,在体液免疫开始时变得可检测到,有可能导致急性情况下病毒学清除延迟。
在任何一例2019冠状病毒病(COVID-19)病例中出现 的严重急性呼吸综合征冠状病毒2(SARS-CoV-2)突变序列理论上可使病毒逃避针对主要感染病毒序列的免疫反应或抗病毒治疗。然而,常用的测序技术并非为检测每个样本中的变异病毒序列而优化设计。为解决这一问题,我们开发了新技术,用于对编码病毒表面蛋白区域的大量个体SARS-CoV-2基因组RNA分子进行测序。这项技术揭示了来自SARS-CoV-2临床分离株的培养病毒中广泛的遗传多样性,但来自7名COVID-19个体的样本中多样性较低。重要的是,对选定个体的配对血清样本进行的同步分析显示,在初始测序时,抗体与SARS-CoV-2刺突蛋白的结合水平相对较低。随着血清与刺突蛋白结合增加,我们检测到在单个表位中有多个携带独立突变的SARS-CoV-2变体,以及病毒载量短暂增加。这些发现表明,SARS-CoV-2复制产生了足够的病毒遗传多样性,以便在急性COVID-19的时间框架内进行免疫介导的变体选择。对SARS-CoV-2变异和特异性免疫反应的大规模研究将有助于确定个体内SARS-CoV-2进化对COVID-19临床结果和抗病毒药物敏感性的影响。