Das Antara, Zhu Bingyao, Xie Yunyao, Zeng Lisha, Pham An T, Neumann Jonathan C, Safrina Olga, Benavides Daniel R, MacGregor Grant R, Schutte Soleil S, Hunt Robert F, O'Dowd Diane K
Department of Developmental and Cell Biology, University of California, Irvine, CA 92697.
Department of Anatomy and Neurobiology, University of California, Irvine, CA 92697.
eNeuro. 2021 Apr 12;8(2). doi: 10.1523/ENEURO.0394-20.2021. Print 2021 Mar-Apr.
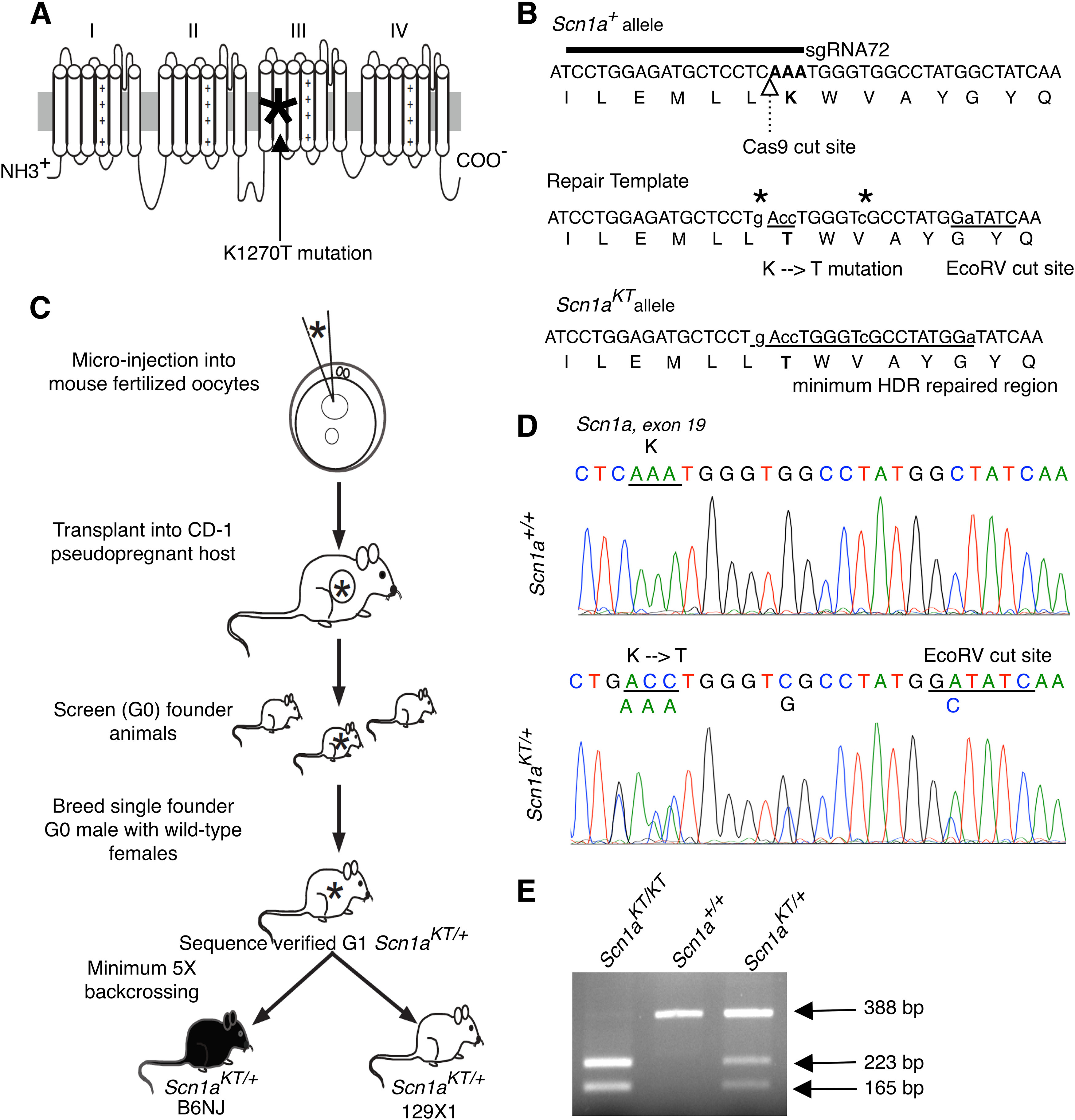
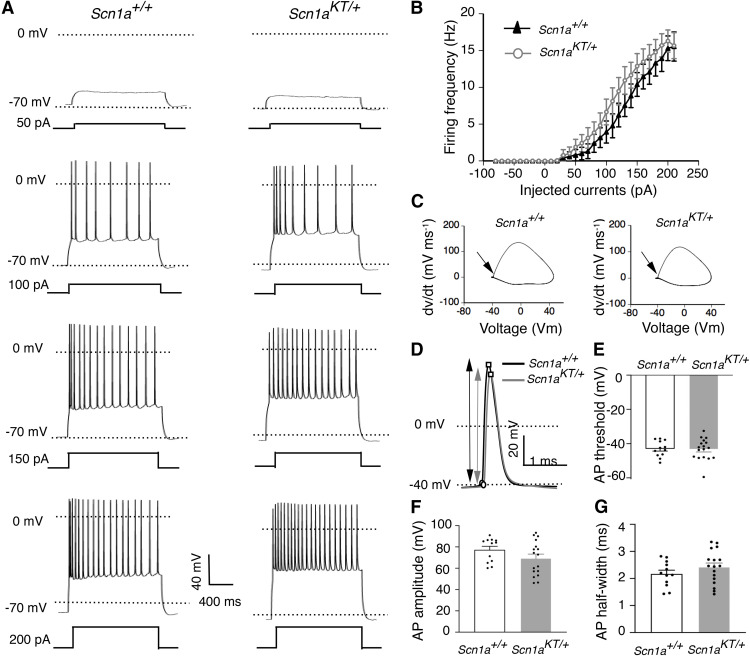
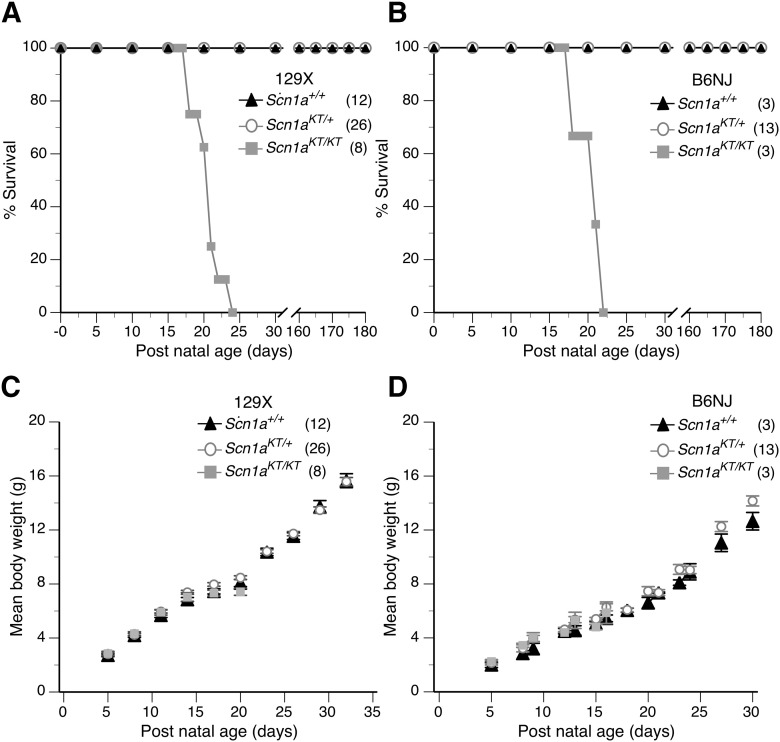
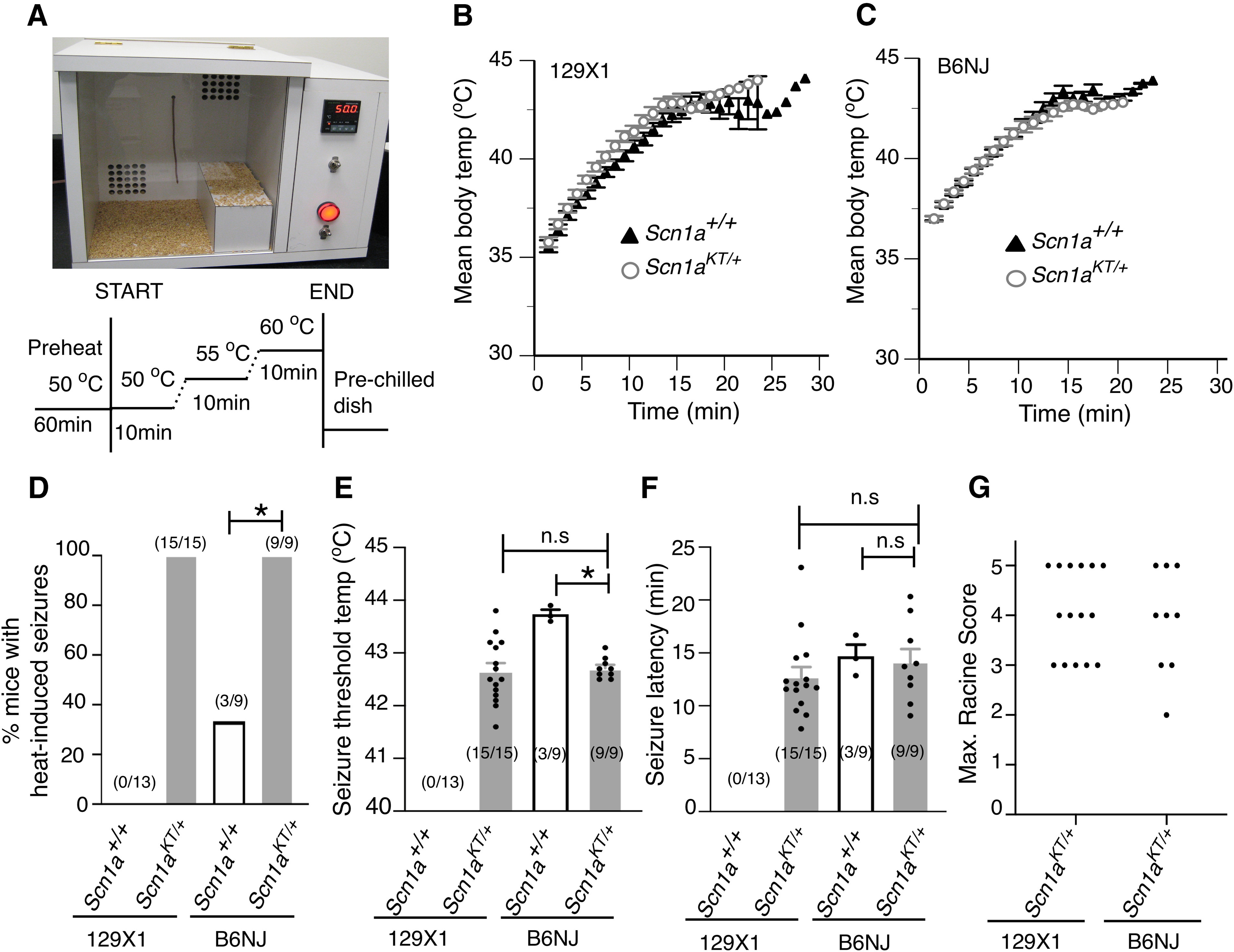
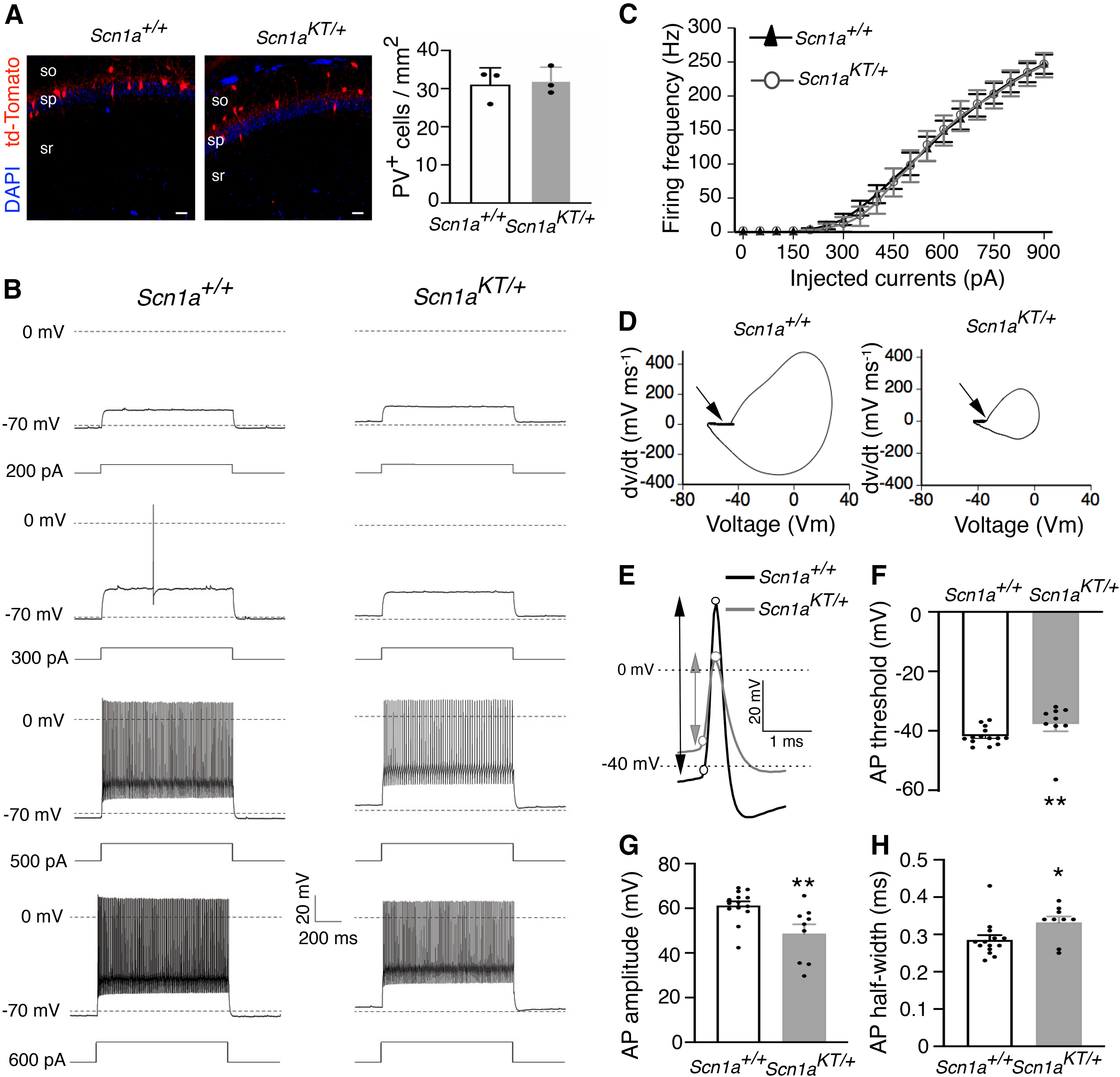
Advances in genome sequencing have identified over 1300 mutations in the sodium channel gene that result in genetic epilepsies. However, it still remains unclear how most individual mutations within result in seizures. A previous study has shown that the K1270T (KT) mutation, linked to genetic epilepsy with febrile seizure plus (GEFS+) in humans, causes heat-induced seizure activity associated with a temperature-dependent decrease in GABAergic neuron excitability in a knock-in model. To examine the behavioral and cellular effects of this mutation in mammals, we introduced the equivalent KT mutation into the mouse () ( gene using CRISPR/Cas9 and generated mutant lines in two widely used genetic backgrounds: C57BL/6NJ and 129X1/SvJ. In both backgrounds, mice homozygous for the KT mutation had spontaneous seizures and died by postnatal day (P)23. There was no difference in mortality of heterozygous KT mice compared with wild-type littermates up to six months old. Heterozygous mutants exhibited heat-induced seizures at ∼42°C, a temperature that did not induce seizures in wild-type littermates. In acute hippocampal slices at permissive temperatures, current-clamp recordings revealed a significantly depolarized shift in action potential threshold and reduced action potential amplitude in parvalbumin (PV)-expressing inhibitory CA1 interneurons in mice. There was no change in the firing properties of excitatory CA1 pyramidal neurons. These results suggest that a constitutive decrease in inhibitory interneuron excitability contributes to the seizure phenotype in the mouse model.
基因组测序技术的进步已在钠通道基因中鉴定出1300多种导致遗传性癫痫的突变。然而,大多数单个突变如何导致癫痫发作仍不清楚。先前的一项研究表明,与人类伴有热性惊厥附加症的遗传性癫痫(GEFS+)相关的K1270T(KT)突变,在一个基因敲入模型中会引起热诱导的癫痫发作活动,同时伴有GABA能神经元兴奋性的温度依赖性降低。为了研究这种突变在哺乳动物中的行为和细胞效应,我们使用CRISPR/Cas9将等效的KT突变引入小鼠()基因,并在两种广泛使用的遗传背景中培育出突变系:C57BL/6NJ和129X1/SvJ。在这两种背景下,KT突变纯合子小鼠均出现自发性癫痫发作,并在出生后第23天死亡。与野生型同窝小鼠相比,杂合子KT小鼠在6个月大之前的死亡率没有差异。杂合子突变体在约42°C时出现热诱导的癫痫发作,而这个温度不会诱导野生型同窝小鼠癫痫发作。在适宜温度下的急性海马切片中,电流钳记录显示,小鼠中表达小白蛋白(PV)的抑制性CA1中间神经元动作电位阈值显著去极化偏移,动作电位幅度降低。兴奋性CA1锥体神经元的放电特性没有变化。这些结果表明,抑制性中间神经元兴奋性的组成性降低导致了小鼠模型中的癫痫发作表型。