Natural and Medical Sciences Research Center, University of Nizwa, Birkat Al-Mouz, Nizwa 616, Sultanate of Oman.
Department of Biosciences, COMSATS University Islamabad, Park Road, Chak Shahzad, Islamabad 45600, Pakistan.
Biomolecules. 2021 Feb 22;11(2):329. doi: 10.3390/biom11020329.
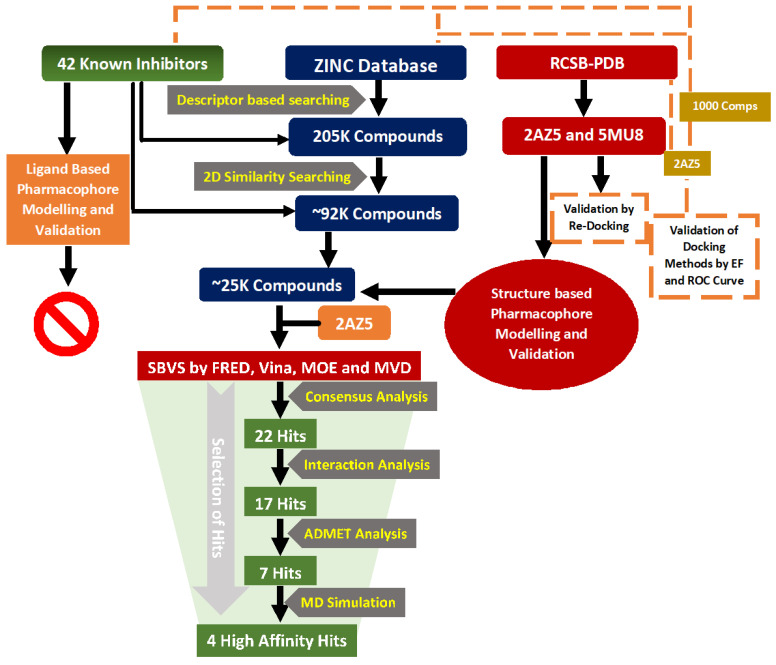
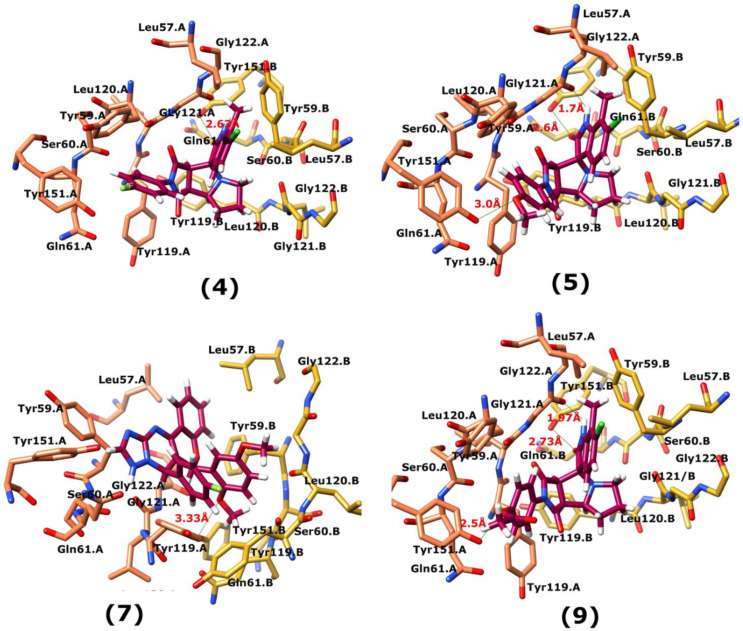
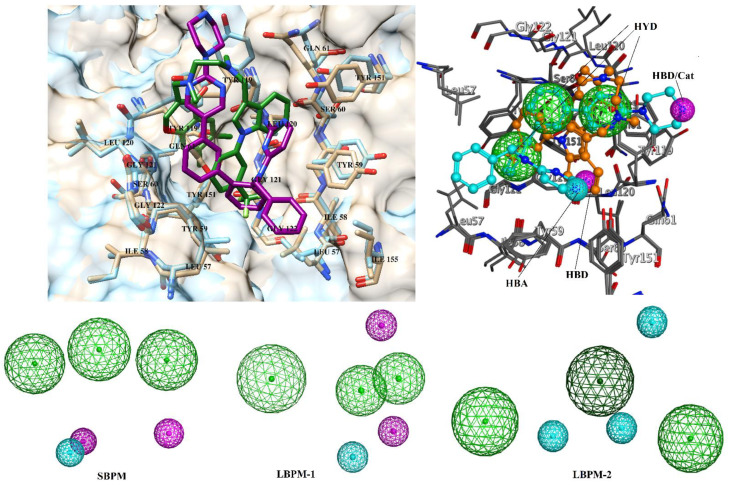
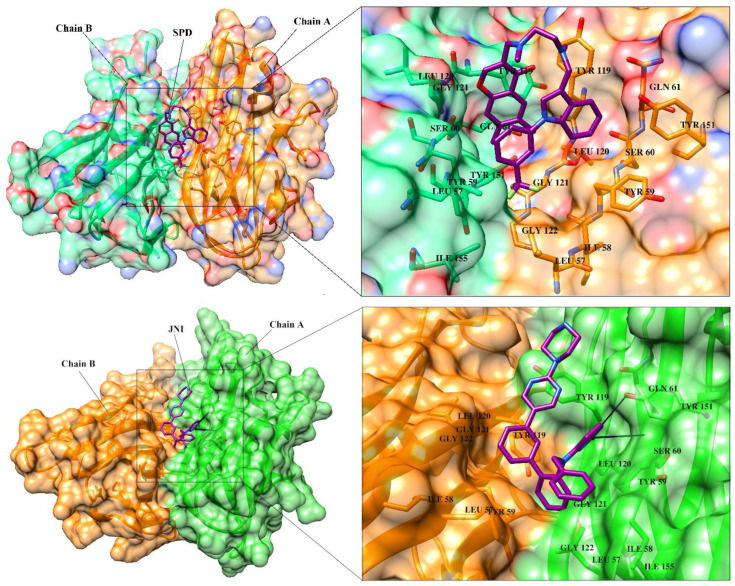
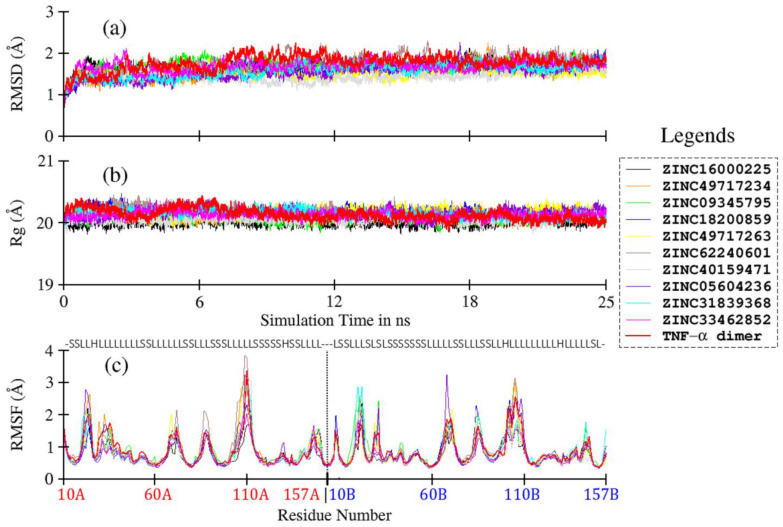
Tumor necrosis factor-α (TNF-α) is a drug target in rheumatoid arthritis and several other auto-immune disorders. TNF-α binds with TNF receptors (TNFR), located on the surface of several immunological cells to exert its effect. Hence, the use of inhibitors that can hinder the complex formation of TNF-α/TNFR can be of medicinal significance. In this study, multiple chem-informatics approaches, including descriptor-based screening, 2D-similarity searching, and pharmacophore modelling were applied to screen new TNF-α inhibitors. Subsequently, multiple-docking protocols were used, and four-fold post-docking results were analyzed by consensus approach. After structure-based virtual screening, seventeen compounds were mutually ranked in top-ranked position by all the docking programs. Those identified hits target TNF-α dimer and effectively block TNF-α/TNFR interface. The predicted pharmacokinetics and physiological properties of the selected hits revealed that, out of seventeen, seven compounds () possessed excellent ADMET profile. These seven compounds plus three more molecules () were chosen for molecular dynamics simulation studies to probe into ligand-induced structural and dynamic behavior of TNF-α, followed by ligand-TNF-α binding free energy calculation using MM-PBSA. The MM-PBSA calculations revealed that compounds and possess highest affinity for TNF-α; 8, 11, 13-15 exhibited moderate affinities, while compound showed weaker binding affinity with TNF-α. This study provides valuable insights to design more potent and selective inhibitors of TNF-α, that will help to treat inflammatory disorders.
肿瘤坏死因子-α(TNF-α)是类风湿关节炎和几种其他自身免疫性疾病的药物靶点。TNF-α与位于几种免疫细胞表面的 TNF 受体(TNFR)结合,发挥其作用。因此,使用可以阻碍 TNF-α/TNFR 复合物形成的抑制剂可能具有医学意义。在这项研究中,应用了多种化学信息学方法,包括基于描述符的筛选、2D 相似性搜索和药效团建模,以筛选新的 TNF-α 抑制剂。随后,使用了多种对接方案,并通过共识方法分析了四轮对接后的结果。基于结构的虚拟筛选后,所有对接程序都将 17 种化合物相互排名在首位。这些鉴定的命中靶标 TNF-α 二聚体,并有效地阻断 TNF-α/TNFR 界面。所选命中物的预测药代动力学和生理特性表明,在这 17 种化合物中,有 7 种()具有良好的 ADMET 特性。这七种化合物加上另外三种分子()被选中进行分子动力学模拟研究,以探究配体诱导的 TNF-α的结构和动态行为,然后使用 MM-PBSA 计算配体-TNF-α的结合自由能。MM-PBSA 计算表明,化合物和具有与 TNF-α 最高的亲和力;化合物 8、11、13-15 表现出中等亲和力,而化合物 与 TNF-α 的结合亲和力较弱。这项研究为设计更有效和更具选择性的 TNF-α抑制剂提供了有价值的见解,这将有助于治疗炎症性疾病。