Huang Chunling, Yi Hao, Shi Ying, Cao Qinghua, Shi Yin, Cheng Delfine, Braet Filip, Chen Xin-Ming, Pollock Carol A
Kolling Institute, Sydney Medical School Northern, Faculty of Medicine and Health, University of Sydney, Royal North Shore Hospital, Sydney, NSW, Australia.
Division of Nephrology, School of Medicine, Stanford University, Stanford, CA, United States.
Front Cell Dev Biol. 2021 Feb 19;9:573814. doi: 10.3389/fcell.2021.573814. eCollection 2021.
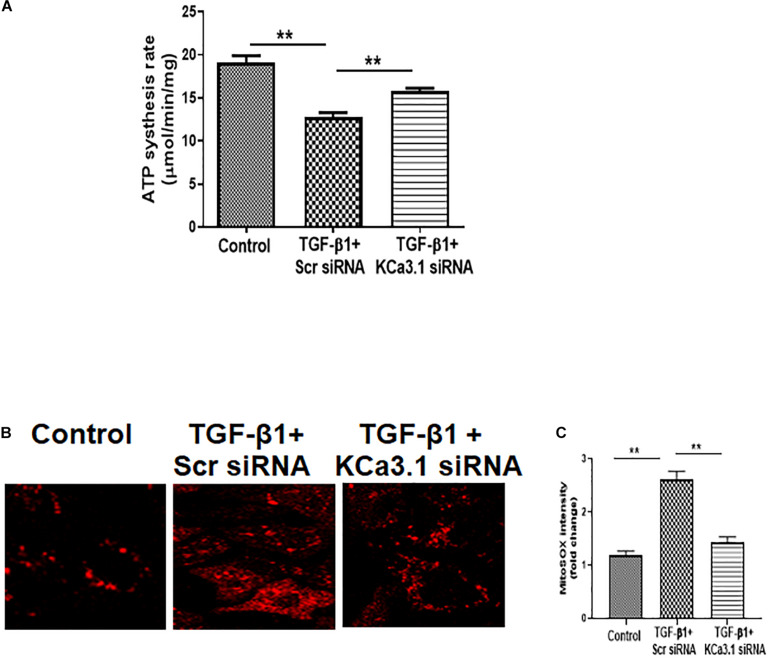
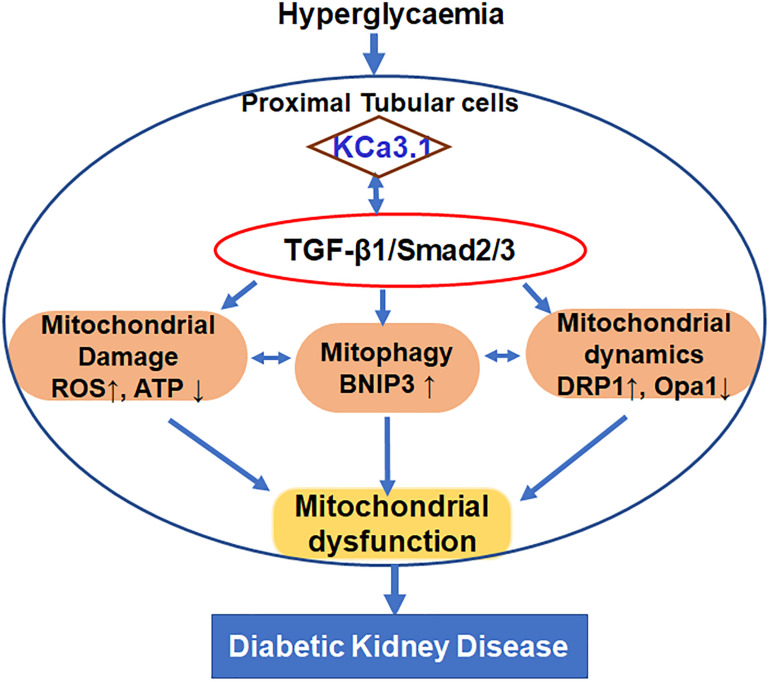
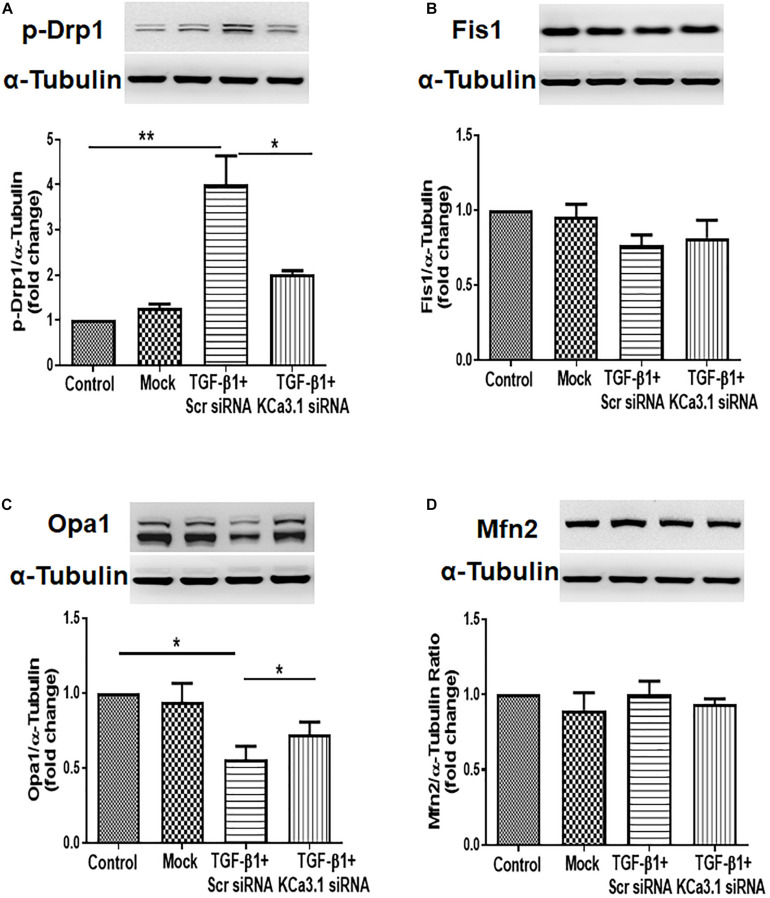
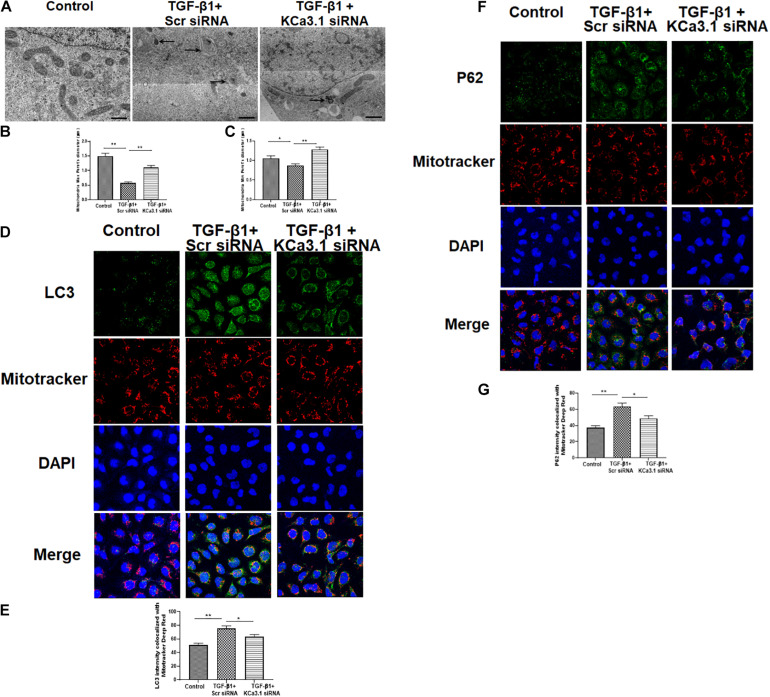
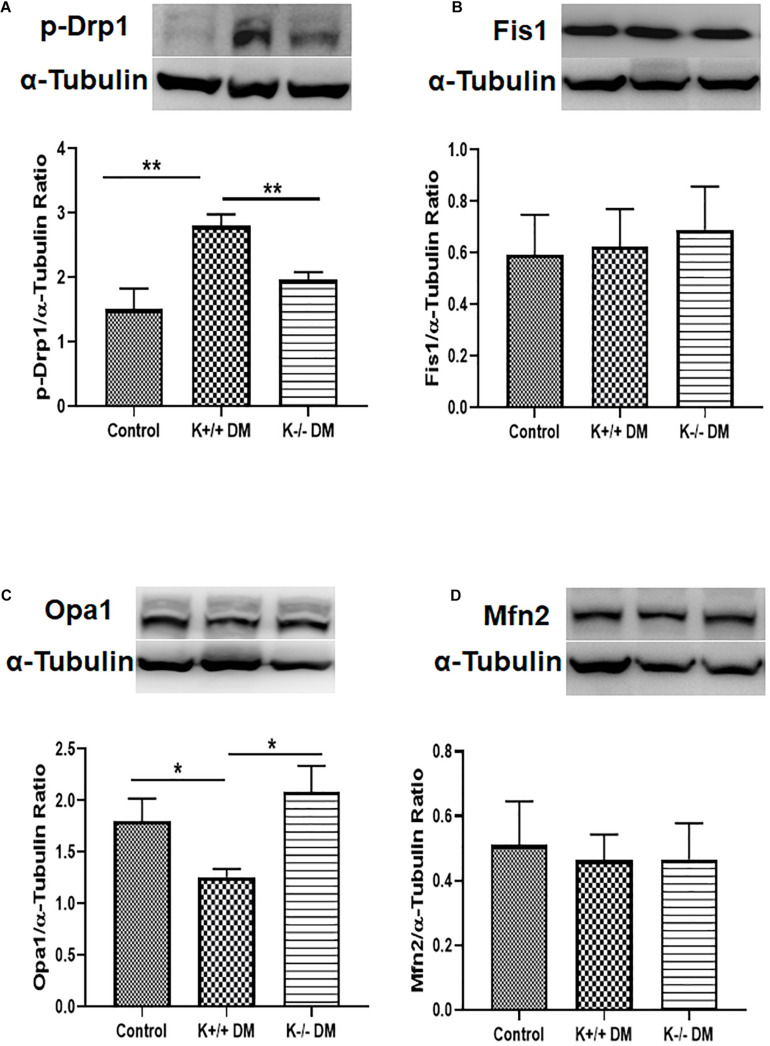
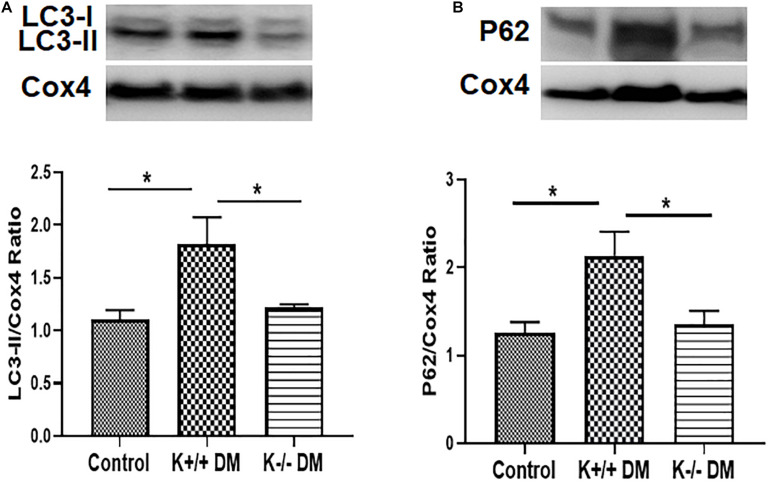
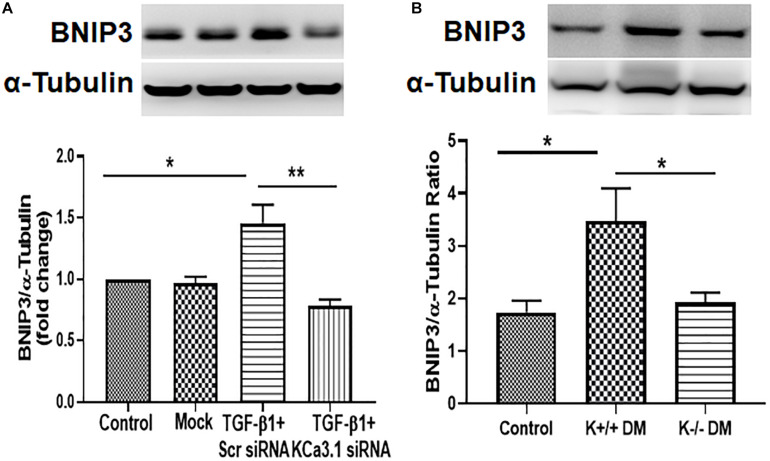
Mitochondrial dysfunction is implicated in the pathogenesis of diabetic kidney disease. Mitochondrial quality control is primarily mediated by mitochondrial turnover and repair through mitochondrial fission/fusion and mitophagy. We have previously shown that blockade of the calcium-activated potassium channel KCa3.1 ameliorates diabetic renal fibrosis. However, the mechanistic link between KCa3.1 and mitochondrial quality control in diabetic kidney disease is not yet known. Transforming growth factor β1 (TGF-β1) plays a central role in diabetic kidney disease. Recent studies indicate an emerging role of TGF-β1 in the regulation of mitochondrial function. However, the molecular mechanism mediating mitochondrial quality control in response to TGF-β1 remains limited. In this study, mitochondrial function was assessed in TGF-β1-exposed renal proximal tubular epithelial cells (HK2 cells) transfected with scrambled siRNA or KCa3.1 siRNA. , diabetes was induced in KCa3.1+/+ and KCa3.1-/- mice by low-dose streptozotocin (STZ) injection. Mitochondrial fission/fusion-related proteins and mitophagy markers, as well as BCL2 interacting protein 3 (BNIP3) (a mitophagy regulator) were examined in HK2 cells and diabetic mice kidneys. The results showed that TGF-β1 significantly inhibited mitochondrial ATP production rate and increased mitochondrial ROS (mtROS) production when compared to control, which was normalized by KCa3.1 gene silencing. Increased fission and suppressed fusion were found in both TGF-β1-treated HK2 cells and diabetic mice, which were reversed by KCa3.1 deficiency. Furthermore, our results showed that mitophagy was inhibited in both and models of diabetic kidney disease. KCa3.1 deficiency restored abnormal mitophagy by inhibiting BNIP3 expression in TGF-β1-induced HK2 cells as well as in the diabetic mice. Collectively, these results indicate that KCa3.1 mediates the dysregulation of mitochondrial quality control in diabetic kidney disease.
线粒体功能障碍与糖尿病肾病的发病机制有关。线粒体质量控制主要通过线粒体分裂/融合和线粒体自噬介导的线粒体更新和修复来实现。我们之前已经表明,钙激活钾通道KCa3.1的阻断可改善糖尿病肾纤维化。然而,在糖尿病肾病中,KCa3.1与线粒体质量控制之间的机制联系尚不清楚。转化生长因子β1(TGF-β1)在糖尿病肾病中起核心作用。最近的研究表明,TGF-β1在调节线粒体功能方面有新的作用。然而,介导对TGF-β1反应的线粒体质量控制的分子机制仍然有限。在本研究中,在用乱序siRNA或KCa3.1 siRNA转染的TGF-β1处理的肾近端小管上皮细胞(HK2细胞)中评估线粒体功能。通过低剂量链脲佐菌素(STZ)注射在KCa3.1+/+和KCa3.1-/-小鼠中诱导糖尿病。在HK2细胞和糖尿病小鼠肾脏中检测线粒体分裂/融合相关蛋白和线粒体自噬标志物,以及BCL2相互作用蛋白3(BNIP3)(一种线粒体自噬调节因子)。结果表明,与对照相比,TGF-β1显著抑制线粒体ATP产生率并增加线粒体ROS(mtROS)产生,而KCa3.1基因沉默可使其恢复正常。在TGF-β1处理的HK2细胞和糖尿病小鼠中均发现裂变增加和融合受抑制,而KCa3.1缺乏可使其逆转。此外,我们的结果表明,在糖尿病肾病的两种模型中,线粒体自噬均受到抑制。KCa3.1缺乏通过抑制TGF-β1诱导的HK2细胞以及糖尿病小鼠中的BNIP3表达来恢复异常的线粒体自噬。总的来说,这些结果表明KCa3.1介导糖尿病肾病中线粒体质量控制的失调。