Haldane Allan, Levy Ronald M
Center for Biophysics and Computational Biology and Department of Physics, Temple University, Philadelphia, Pennsylvania 19122.
Center for Biophysics and Computational Biology and Department of Chemistry, Temple University, Philadelphia, Pennsylvania 19122.
Comput Phys Commun. 2021 Mar;260. doi: 10.1016/j.cpc.2020.107312. Epub 2020 Apr 17.
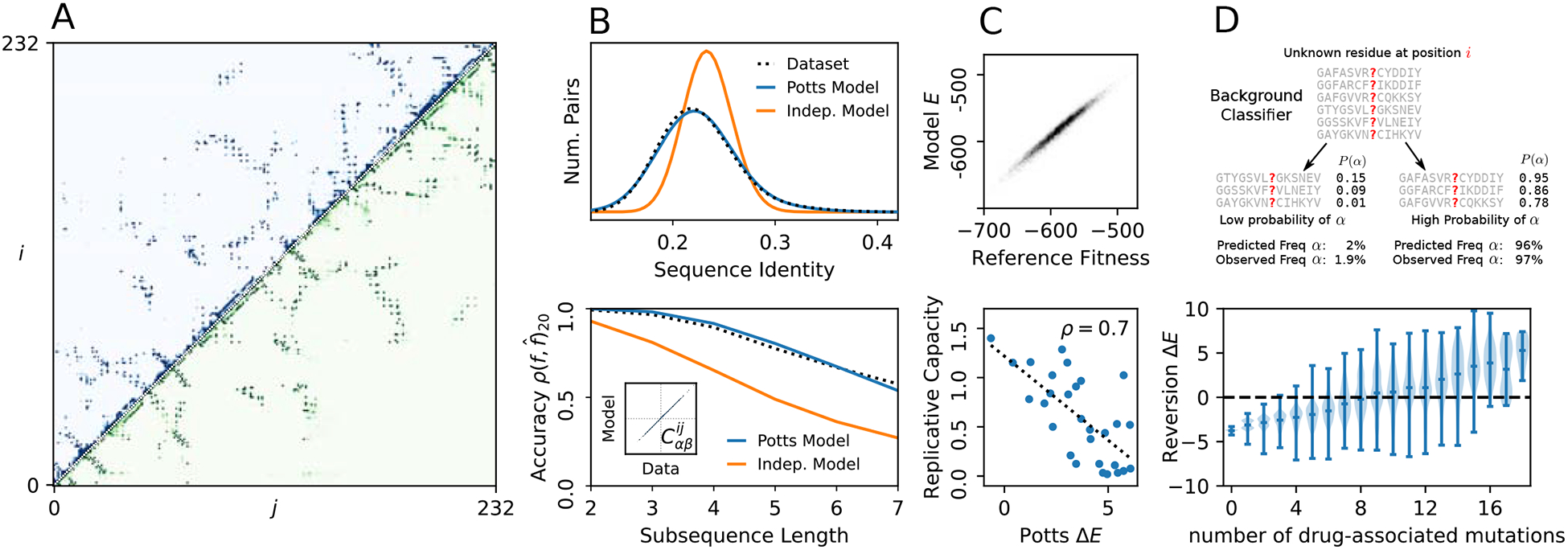
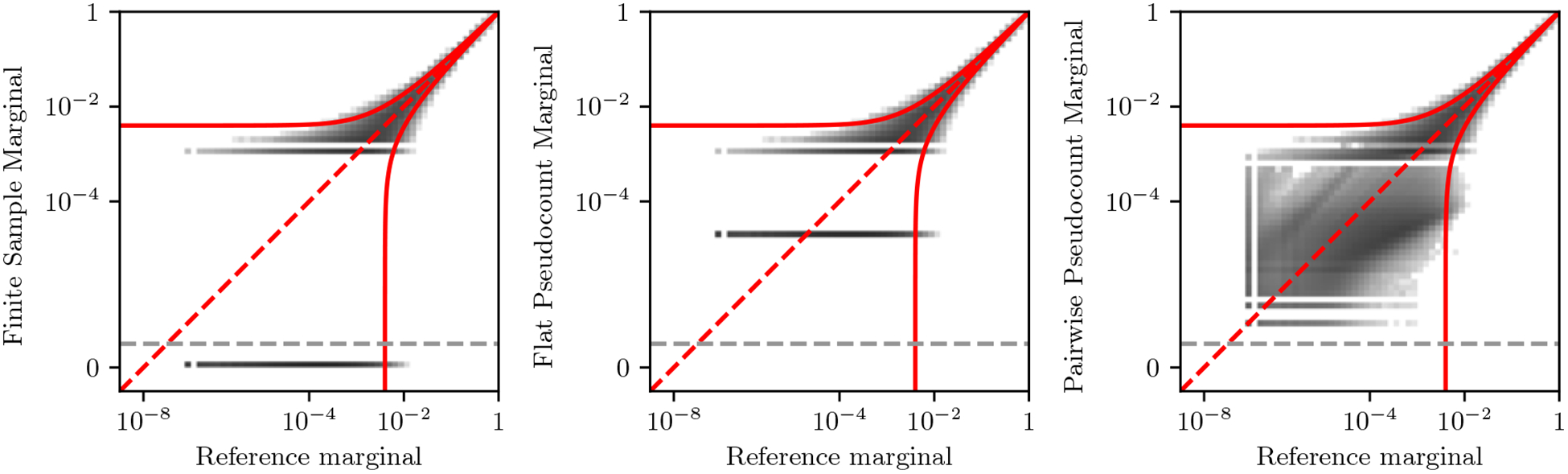
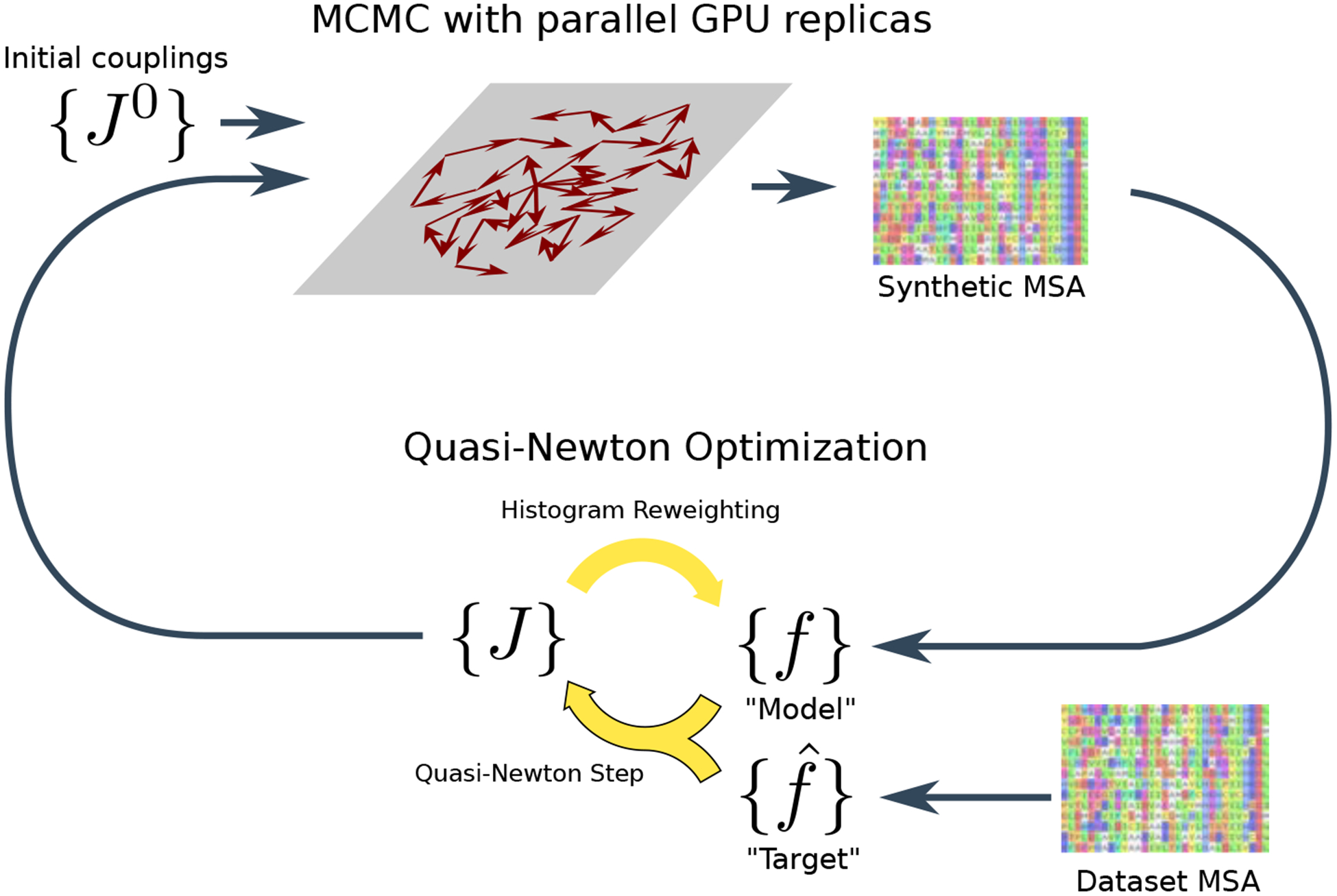
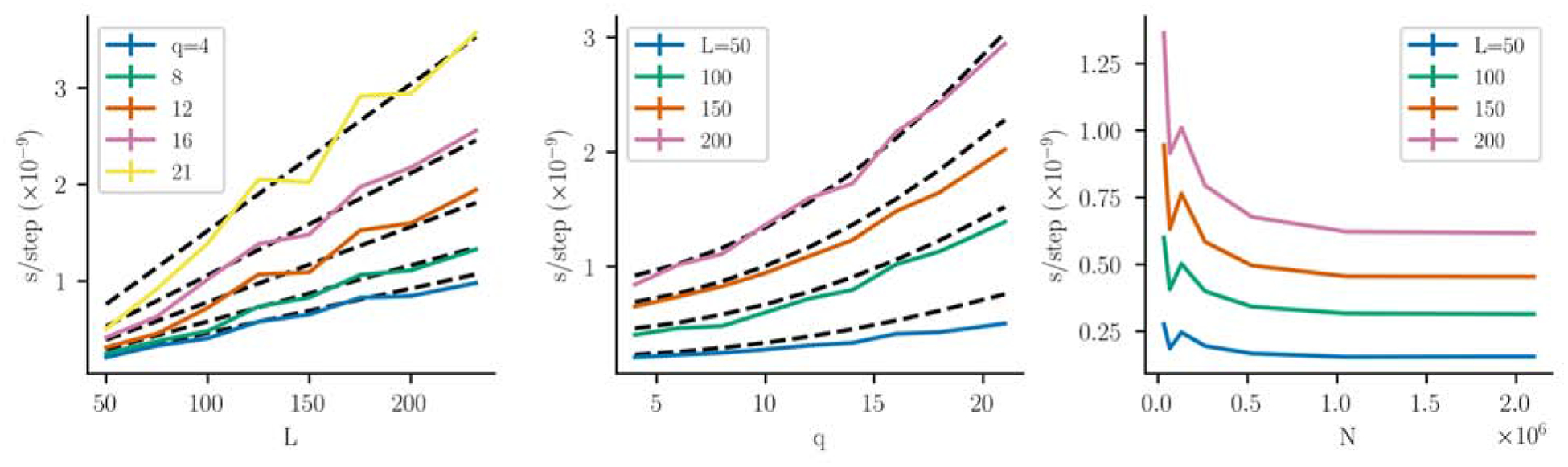
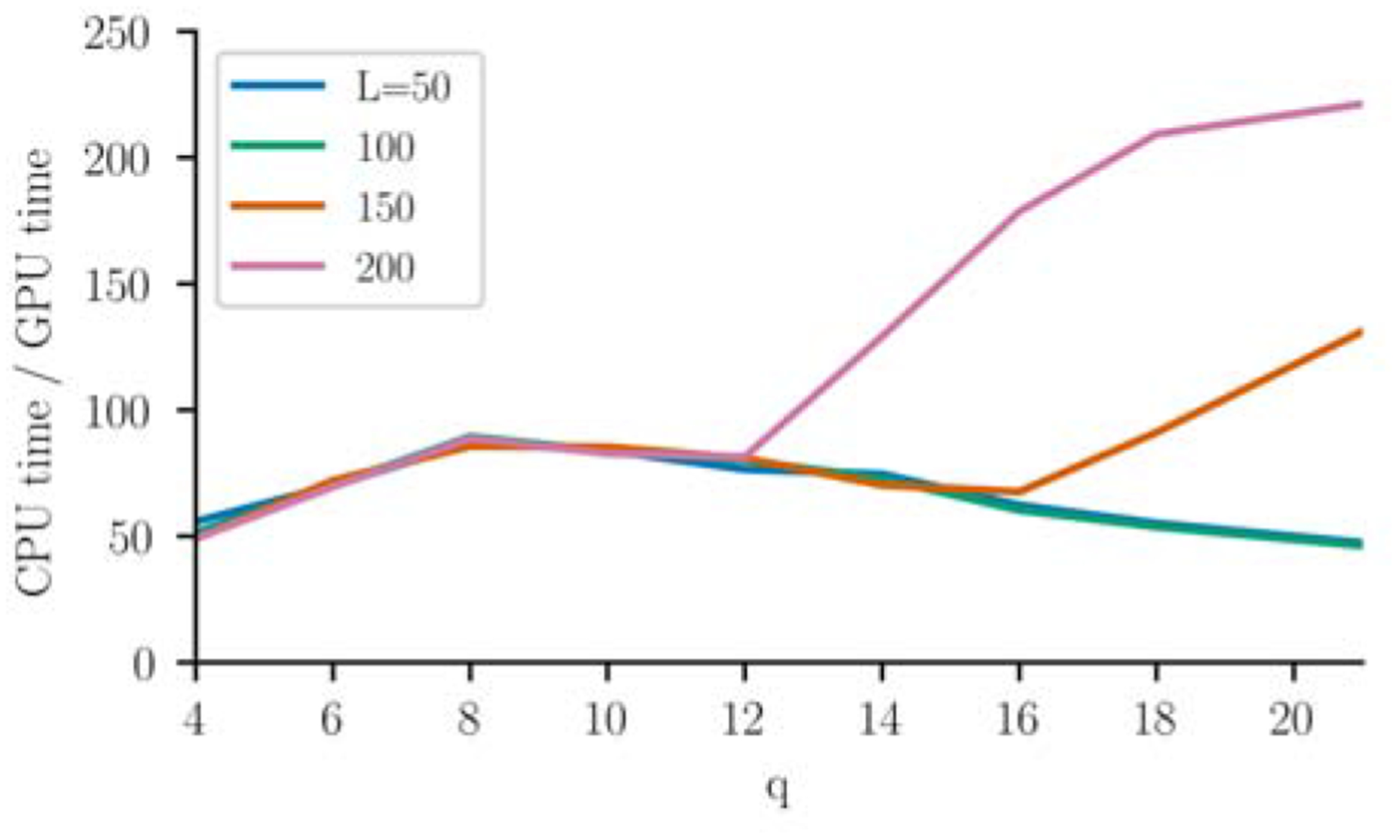
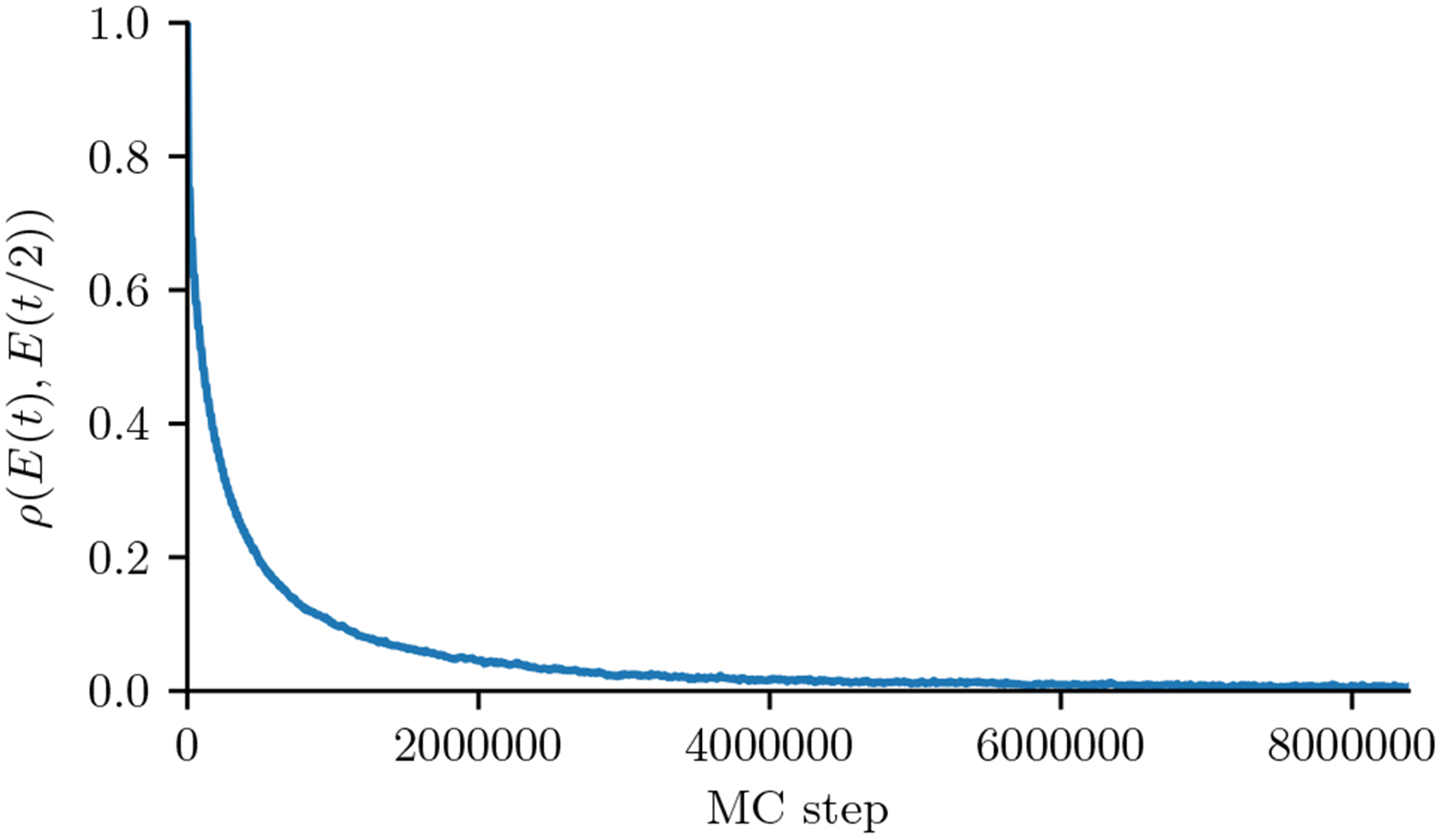
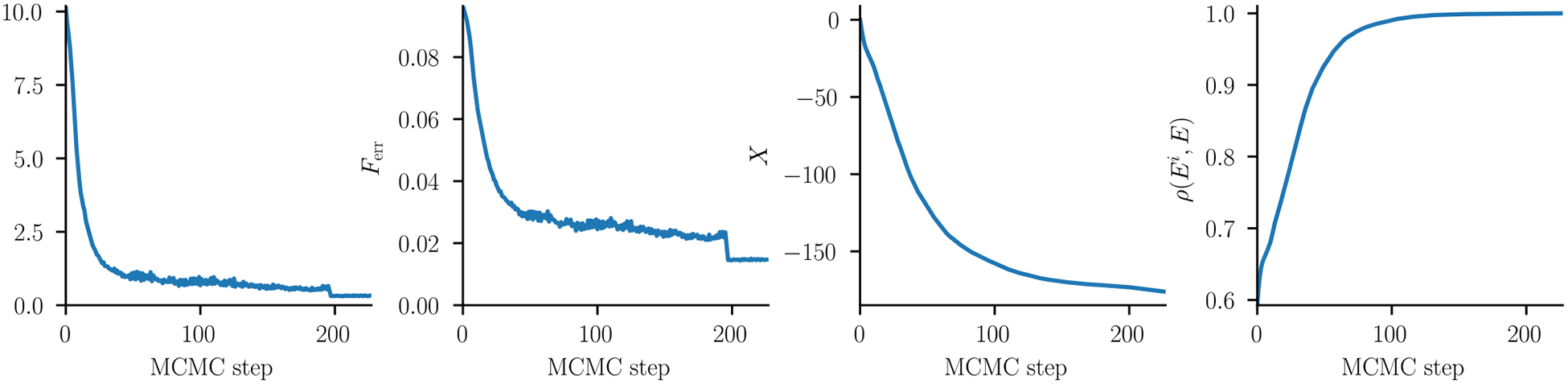
Inverse Ising inference is a method for inferring the coupling parameters of a Potts/Ising model based on observed site-covariation, which has found important applications in protein physics for detecting interactions between residues in protein families. We introduce Mi3-GPU ("mee-three", for CMC nverse sing nference) software for solving the inverse Ising problem for protein-sequence datasets with few analytic approximations, by parallel Markov-Chain Monte-Carlo sampling on GPUs. We also provide tools for analysis and preparation of protein-family Multiple Sequence Alignments (MSAs) to account for finite-sampling issues, which are a major source of error or bias in inverse Ising inference. Our method is "generative" in the sense that the inferred model can be used to generate synthetic MSAs whose mutational statistics (marginals) can be verified to match the dataset MSA statistics up to the limits imposed by the effects of finite sampling. Our GPU implementation enables the construction of models which reproduce the covariation patterns of the observed MSA with a precision that is not possible with more approximate methods. The main components of our method are a GPU-optimized algorithm to greatly accelerate MCMC sampling, combined with a multi-step Quasi-Newton parameter-update scheme using a "Zwanzig reweighting" technique. We demonstrate the ability of this software to produce generative models on typical protein family datasets for sequence lengths ~ 300 with 21 residue types with tens of millions of inferred parameters in short running times.
逆伊辛推理是一种基于观测到的位点协变来推断Potts/伊辛模型耦合参数的方法,它在蛋白质物理学中已被发现可用于检测蛋白质家族中残基之间的相互作用。我们引入了Mi3-GPU(“mee-three”,用于CMC逆伊辛推理)软件,通过在GPU上进行并行马尔可夫链蒙特卡罗采样,以很少的解析近似来解决蛋白质序列数据集的逆伊辛问题。我们还提供了用于分析和准备蛋白质家族多序列比对(MSA)的工具,以解决有限采样问题,有限采样问题是逆伊辛推理中误差或偏差的主要来源。我们的方法是“生成式”的,即推断出的模型可用于生成合成MSA,其突变统计(边际)可以被验证,在有限采样效应所施加的限制范围内,与数据集MSA统计相匹配。我们的GPU实现能够构建出以更近似方法无法达到的精度重现观测到的MSA协变模式的模型。我们方法的主要组成部分是一种GPU优化算法,用于极大地加速MCMC采样,并结合使用“Zwanzig重加权”技术的多步拟牛顿参数更新方案。我们展示了该软件在典型蛋白质家族数据集上生成生成式模型的能力,这些数据集序列长度约为300,有21种残基类型,在短运行时间内有多达数千万个推断参数。