Department of Theoretical Biophysics, Max-Planck-Institute of Biophysics, Frankfurt am Main 60438, Germany.
J Chem Theory Comput. 2021 Apr 13;17(4):2530-2540. doi: 10.1021/acs.jctc.0c01281. Epub 2021 Mar 15.
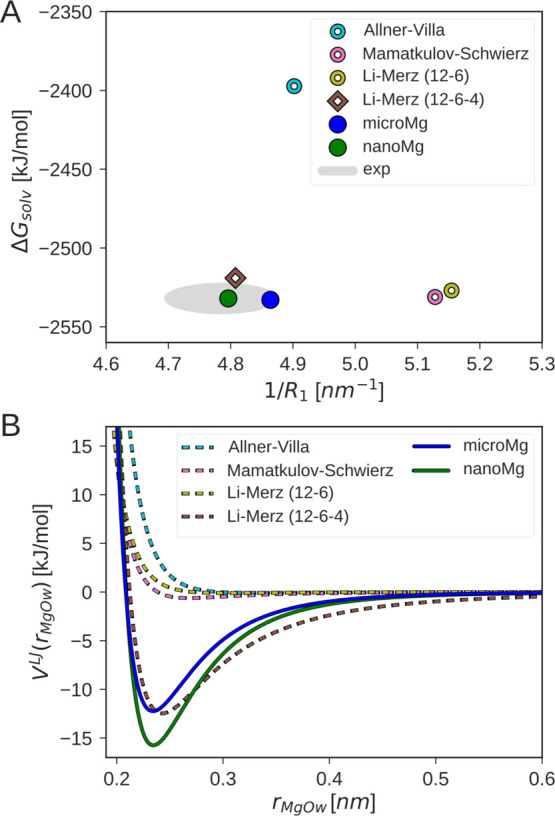
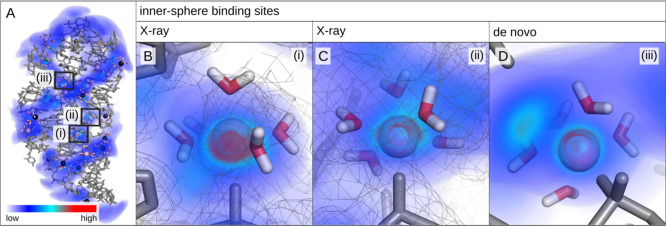
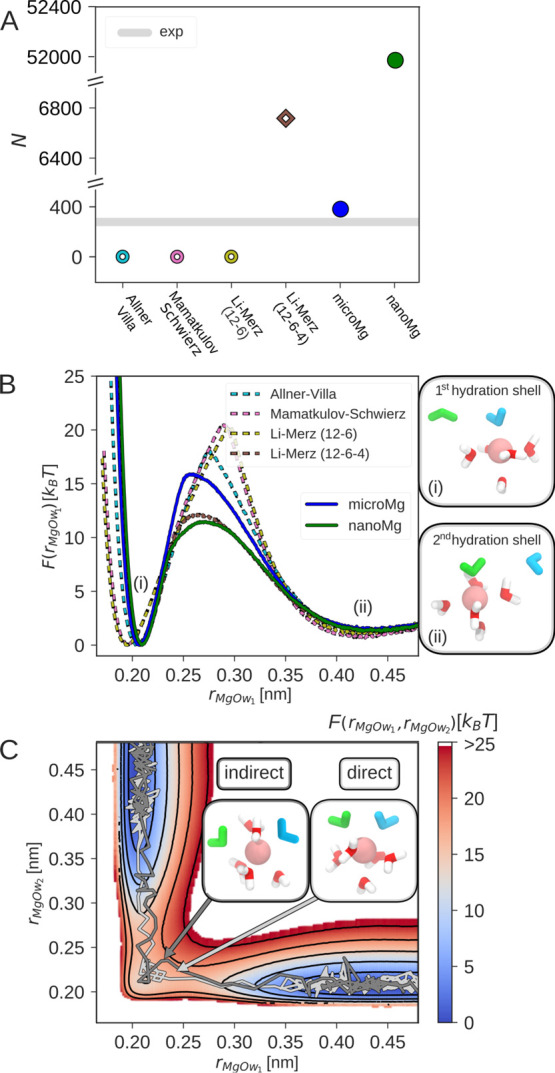
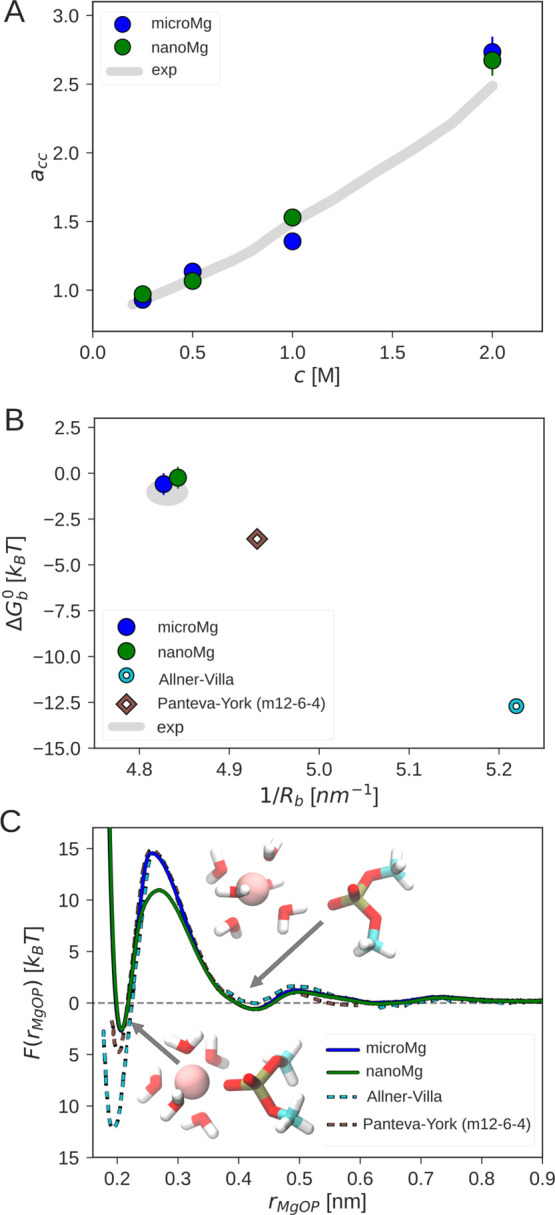
Magnesium ions play an essential role in many vital processes. To correctly describe their interactions in molecular dynamics simulations, an accurate parametrization is crucial. Despite the importance and considerable scientific effort, current force fields based on the commonly used 12-6 Lennard-Jones interaction potential fail to reproduce a variety of experimental solution properties. In particular, no parametrization exists so far that simultaneously reproduces the solvation free energy and the distance to the water oxygens in the first hydration shell. Moreover, current Mg force fields significantly underestimate the rate of water exchange leading to unrealistically slow exchange kinetics. In order to make progress in the development of improved models, we systematically optimize the Mg parameters in combination with the TIP3P water model in a much larger parameter space than previously done. The results show that a long-ranged interaction potential and modified Lorentz-Berthelot combination rules allow us to accurately reproduce multiple experimental properties including the solvation free energy, the distances to the oxygens of the first hydration shell, the hydration number, the activity coefficient derivative in MgCl solutions, the self-diffusion coefficient, and the binding affinity to the phosphate oxygen of RNA. Matching this broad range of thermodynamic properties, we present two sets of optimal parameters: yields water exchange on the microsecond timescale in agreement with experiments. yields water exchange on the nanosecond timescale facilitating the direct observation of ion-binding events. As shown for the example of the A-riboswitch, the optimized parameters correctly reproduce the structure of specifically bound ions and permit the de novo prediction of Mg-binding sites in biomolecular simulations.
镁离子在许多重要的生理过程中起着至关重要的作用。为了在分子动力学模拟中正确描述它们的相互作用,精确的参数化是至关重要的。尽管重要性和相当大的科学努力,目前基于常用的 12-6 Lennard-Jones 相互作用势的力场无法重现各种实验溶液性质。特别是,目前还没有一个参数化可以同时重现溶剂化自由能和第一水合壳层中与水分子氧的距离。此外,目前的 Mg 力场大大低估了水交换的速率,导致不现实的慢交换动力学。为了在改进模型的发展方面取得进展,我们系统地优化了 Mg 参数,同时结合 TIP3P 水模型,在比以前更大的参数空间中进行优化。结果表明,长程相互作用势和修正的 Lorentz-Berthelot 组合规则使我们能够准确地重现多种实验性质,包括溶剂化自由能、第一水合壳层中氧的距离、水合数、MgCl 溶液中的活度系数导数、自扩散系数以及与 RNA 磷酸氧的结合亲和力。匹配这一广泛的热力学性质,我们提出了两套最佳参数:一是使水交换在微秒时间尺度上进行,与实验结果一致;二是使水交换在纳秒时间尺度上进行,有利于直接观察离子结合事件。以 A-核糖开关为例,优化后的参数正确地重现了特定结合离子的结构,并允许在生物分子模拟中从头预测 Mg 结合位点。