Structural Genomics Consortium, Nuffield Department of Medicine, University of Oxford, Oxford, United Kingdom, OX3 7DQ.
Target Discovery Institute, University of Oxford, Oxford, United Kingdom, OX3 7FZ.
ACS Chem Biol. 2021 Apr 16;16(4):586-595. doi: 10.1021/acschembio.0c00498. Epub 2021 Mar 16.
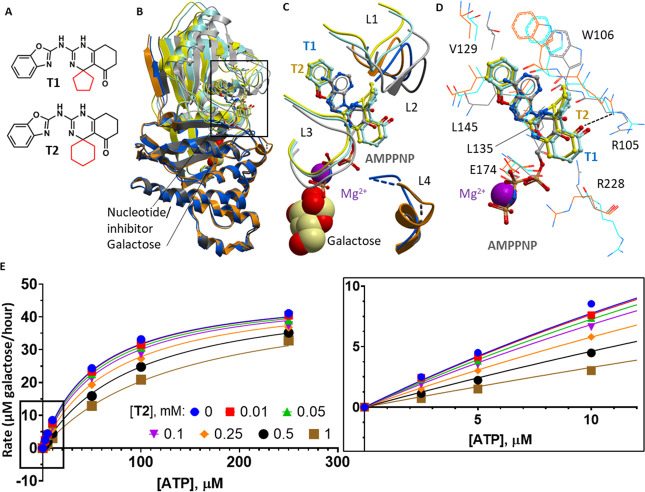
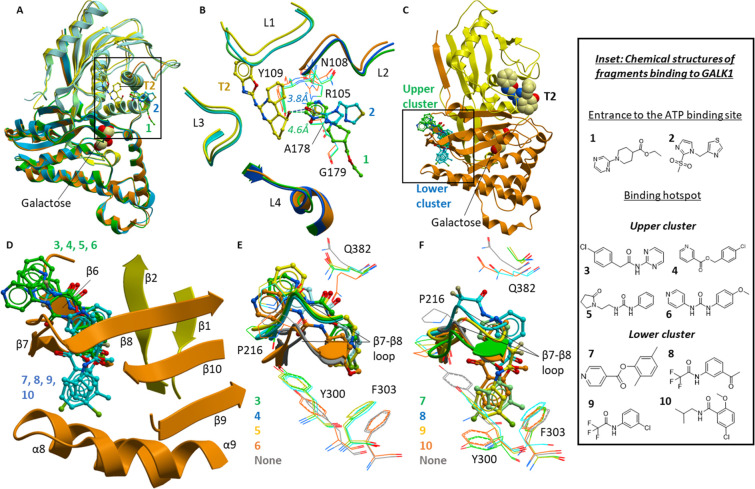
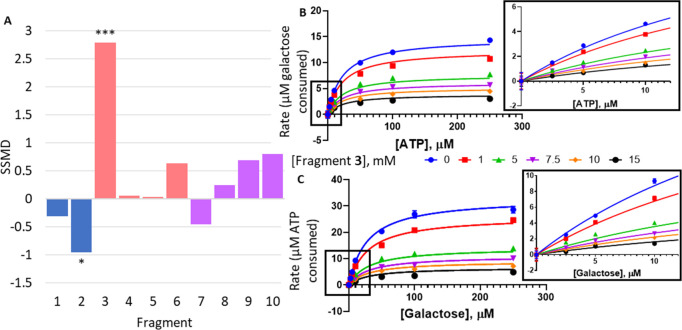
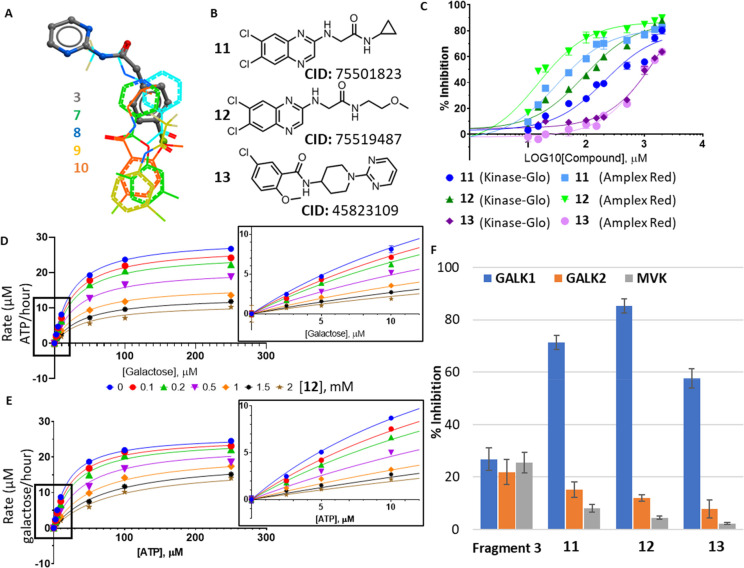
Classic galactosemia is caused by loss-of-function mutations in galactose-1-phosphate uridylyltransferase (GALT) that lead to toxic accumulation of its substrate, galactose-1-phosphate. One proposed therapy is to inhibit the biosynthesis of galactose-1-phosphate, catalyzed by galactokinase 1 (GALK1). Existing inhibitors of human GALK1 (hGALK1) are primarily ATP-competitive with limited clinical utility to date. Here, we determined crystal structures of hGALK1 bound with reported ATP-competitive inhibitors of the spiro-benzoxazole series, to reveal their binding mode in the active site. Spurred by the need for additional chemotypes of hGALK1 inhibitors, desirably targeting a nonorthosteric site, we also performed crystallography-based screening by soaking hundreds of hGALK1 crystals, already containing active site ligands, with fragments from a custom library. Two fragments were found to bind close to the ATP binding site, and a further eight were found in a hotspot distal from the active site, highlighting the strength of this method in identifying previously uncharacterized allosteric sites. To generate inhibitors of improved potency and selectivity targeting the newly identified binding hotspot, new compounds were designed by merging overlapping fragments. This yielded two micromolar inhibitors of hGALK1 that were not competitive with respect to either substrate (ATP or galactose) and demonstrated good selectivity over hGALK1 homologues, galactokinase 2 and mevalonate kinase. Our findings are therefore the first to demonstrate inhibition of hGALK1 from an allosteric site, with potential for further development of potent and selective inhibitors to provide novel therapeutics for classic galactosemia.
经典型半乳糖血症是由于半乳糖-1-磷酸尿苷酰转移酶(GALT)的功能丧失突变引起的,导致其底物半乳糖-1-磷酸的毒性积累。一种提出的治疗方法是抑制半乳糖激酶 1(GALK1)催化的半乳糖-1-磷酸的生物合成。目前用于抑制人 GALK1(hGALK1)的抑制剂主要是与 ATP 竞争的,临床应用有限。在这里,我们确定了与报道的苯并恶唑螺环系列的 ATP 竞争性抑制剂结合的 hGALK1 的晶体结构,以揭示它们在活性部位的结合模式。由于需要针对 hGALK1 抑制剂的其他化学型,理想情况下是针对非变构位点,我们还通过浸泡数百个已经含有活性位点配体的 hGALK1 晶体进行了基于晶体学的筛选,这些晶体用来自定制文库的片段进行浸泡。发现两个片段紧密结合在 ATP 结合位点附近,另外八个片段位于远离活性位点的热点,突出了这种方法在识别以前未表征的变构位点方面的优势。为了生成针对新鉴定的结合热点的改进效力和选择性的抑制剂,通过合并重叠的片段设计了新的化合物。这产生了两种对 hGALK1 具有微摩尔抑制作用的化合物,它们与底物(ATP 或半乳糖)均不具有竞争性,并且对 hGALK1 同源物、半乳糖激酶 2 和甲羟戊酸激酶具有良好的选择性。因此,我们的发现首次证明了通过变构位点抑制 hGALK1,有可能进一步开发出有效的、选择性的抑制剂,为经典型半乳糖血症提供新的治疗方法。