Department of Chemistry and Biochemistry, University of Oklahoma, 101 Stephenson Parkway, Norman, Oklahoma 73019, United States.
School of Informatics and Computing, Indiana University-Purdue University Indianapolis, Indianapolis, Indiana 46202, United States.
J Am Soc Mass Spectrom. 2021 Jun 2;32(6):1336-1344. doi: 10.1021/jasms.0c00464. Epub 2021 Mar 16.
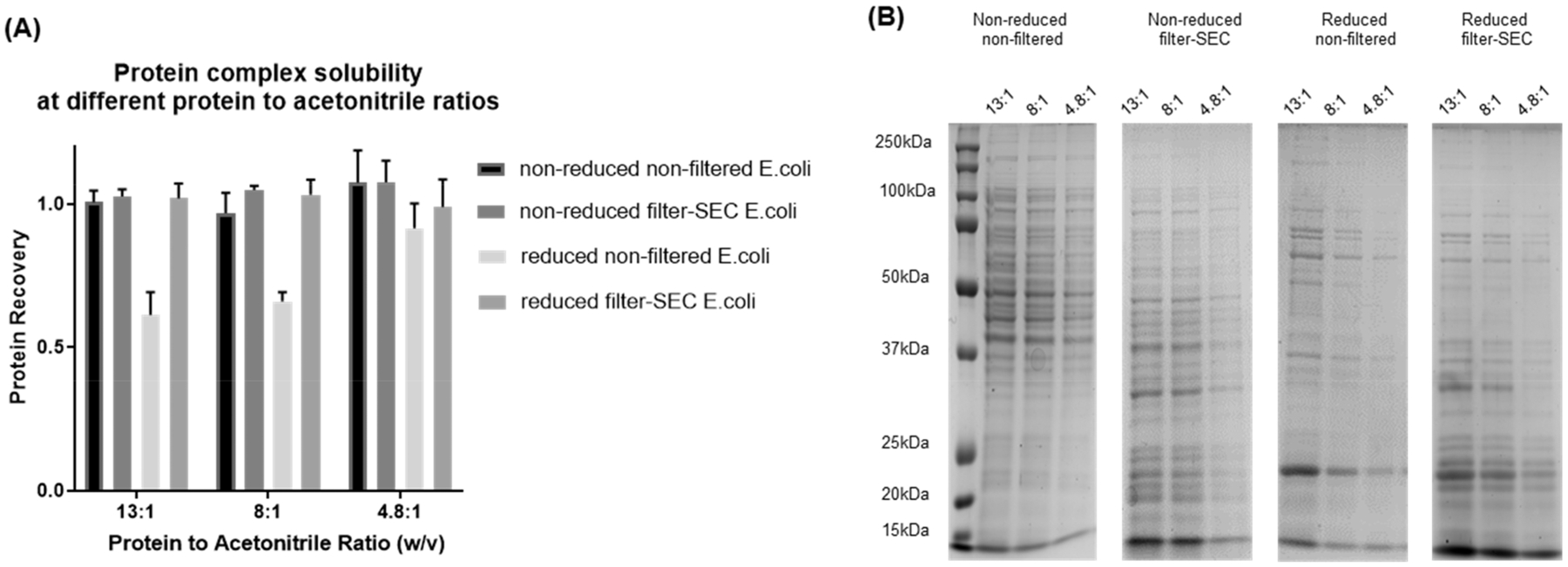
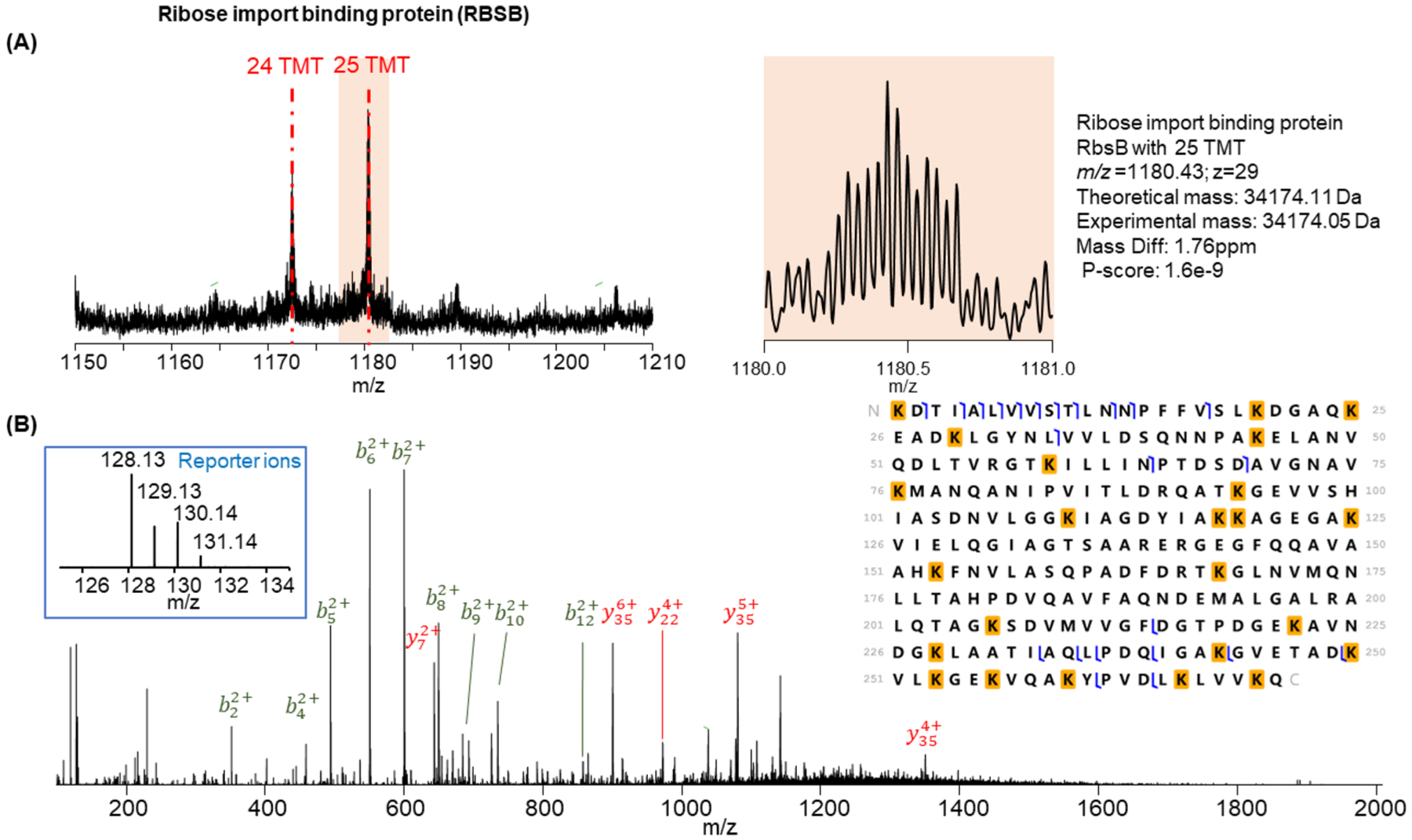
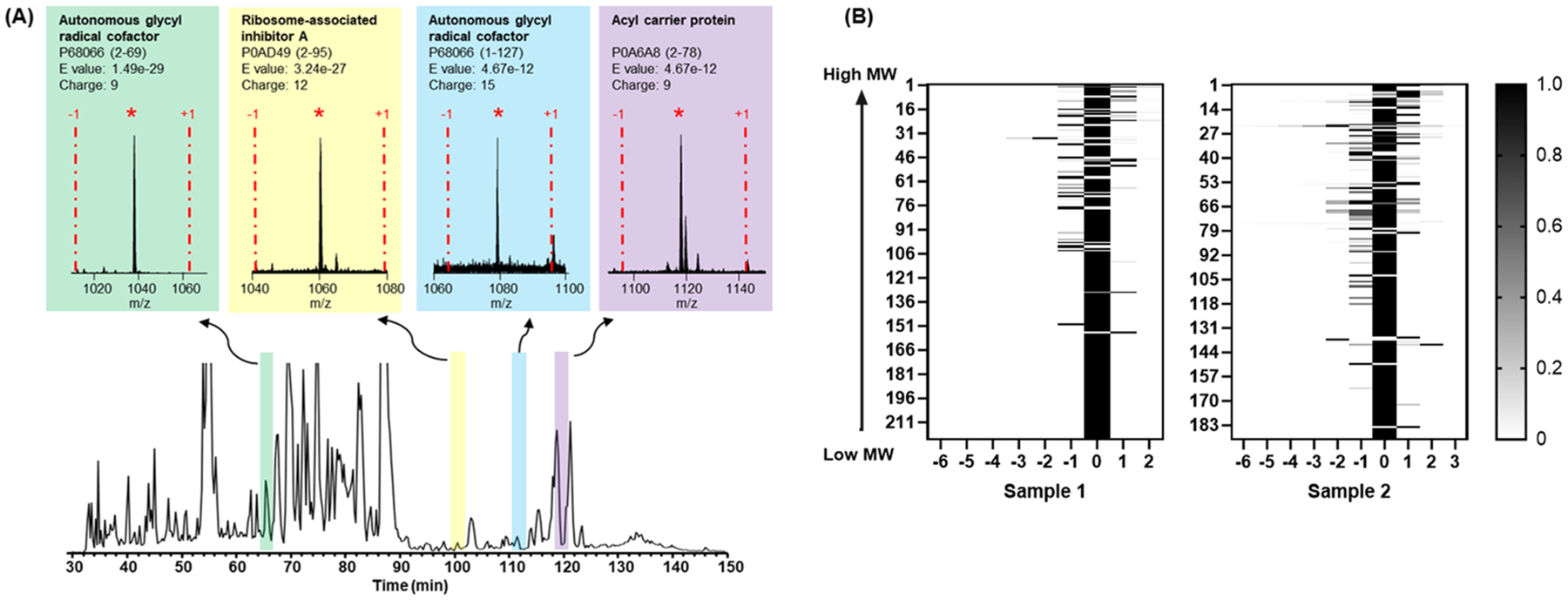
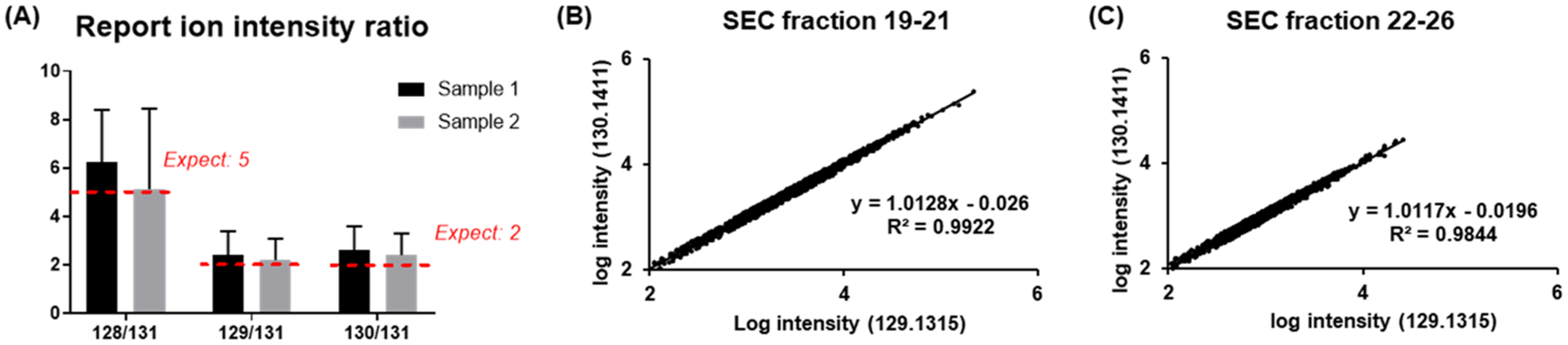
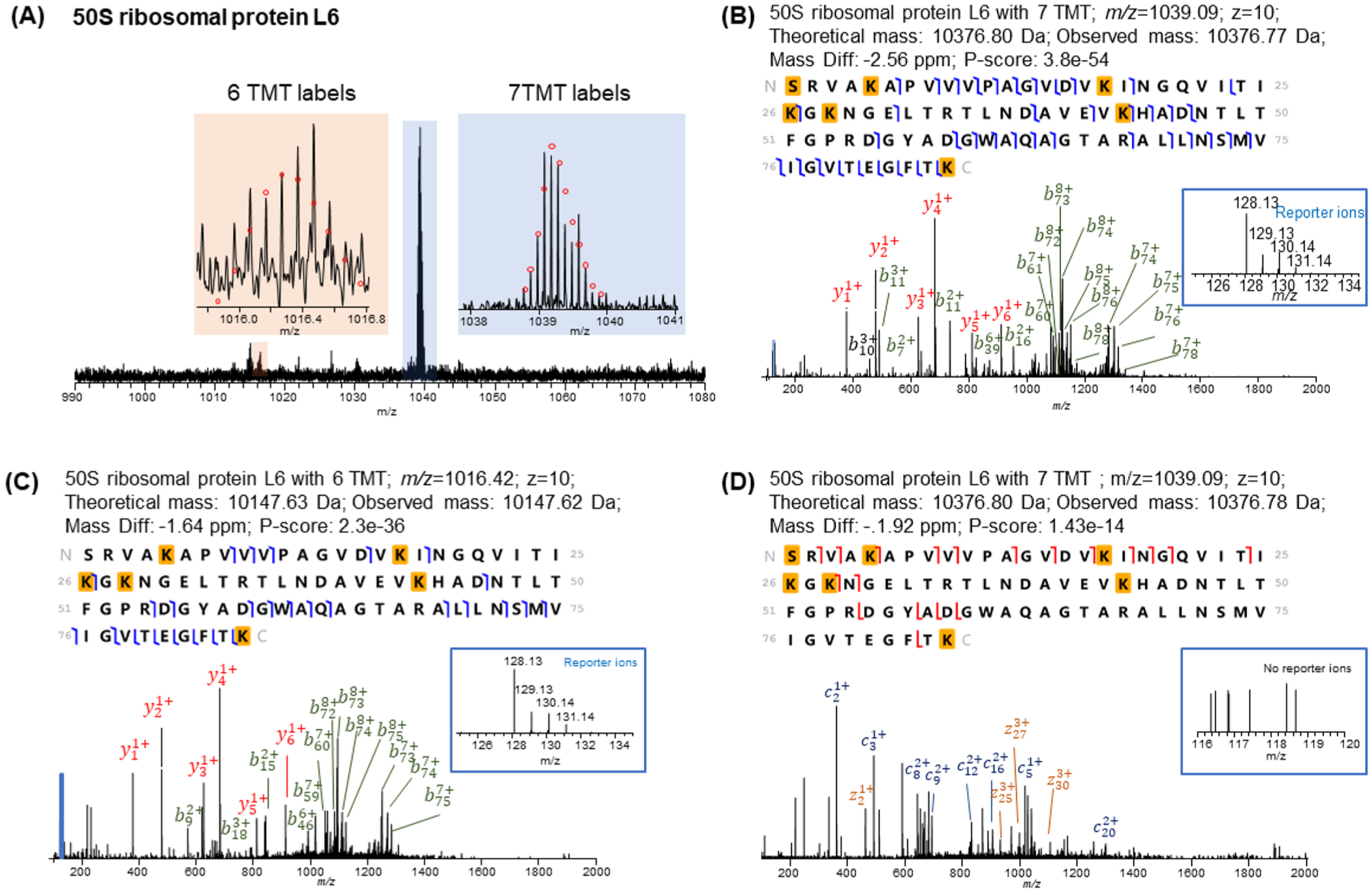
Labeling approaches using isobaric chemical tags (e.g., isobaric tagging for relative and absolute quantification, iTRAQ and tandem mass tag, TMT) have been widely applied for the quantification of peptides and proteins in bottom-up MS. However, until recently, successful applications of these approaches to top-down proteomics have been limited because proteins tend to precipitate and "crash" out of solution during TMT labeling of complex samples making the quantification of such samples difficult. In this study, we report a top-down TMT MS platform for confidently identifying and quantifying low molecular weight intact proteoforms in complex biological samples. To reduce the sample complexity and remove large proteins from complex samples, we developed a filter-SEC technique that combines a molecular weight cutoff filtration step with high-performance size exclusion chromatography (SEC) separation. No protein precipitation was observed in filtered samples under the intact protein-level TMT labeling conditions. The proposed top-down TMT MS platform enables high-throughput analysis of intact proteoforms, allowing for the identification and quantification of hundreds of intact proteoforms from cell lysates. To our knowledge, this represents the first high-throughput TMT labeling-based, quantitative, top-down MS analysis suitable for complex biological samples.
基于等压化学标签(例如,相对和绝对定量的同位素标记、iTRAQ 和串联质量标签、TMT)的标记方法已广泛应用于基于 MS 的肽和蛋白质的定量。然而,直到最近,这些方法在自上而下的蛋白质组学中的成功应用一直受到限制,因为在复杂样品的 TMT 标记过程中,蛋白质往往会沉淀并“崩溃”出溶液,使得此类样品的定量变得困难。在这项研究中,我们报告了一种用于自信地鉴定和定量复杂生物样品中低分子量完整蛋白质形式的自上而下的 TMT MS 平台。为了降低样品复杂性并从复杂样品中去除大蛋白质,我们开发了一种过滤-SEC 技术,该技术将分子量截止过滤步骤与高性能尺寸排阻色谱 (SEC) 分离相结合。在完整蛋白质水平的 TMT 标记条件下,过滤后的样品中未观察到蛋白质沉淀。所提出的自上而下的 TMT MS 平台能够实现完整蛋白质形式的高通量分析,允许从细胞裂解物中鉴定和定量数百种完整蛋白质形式。据我们所知,这代表了第一个适用于复杂生物样品的基于高通量 TMT 标记的定量自上而下的 MS 分析。