Department of Chemistry, Michigan State University, 578 S Shaw Lane, East Lansing, MI, 48824, USA.
Genome Center of Wisconsin, University of Wisconsin-Madison, Madison, WI, 53706, USA.
J Am Soc Mass Spectrom. 2019 Dec;30(12):2470-2479. doi: 10.1007/s13361-019-02206-6. Epub 2019 May 9.
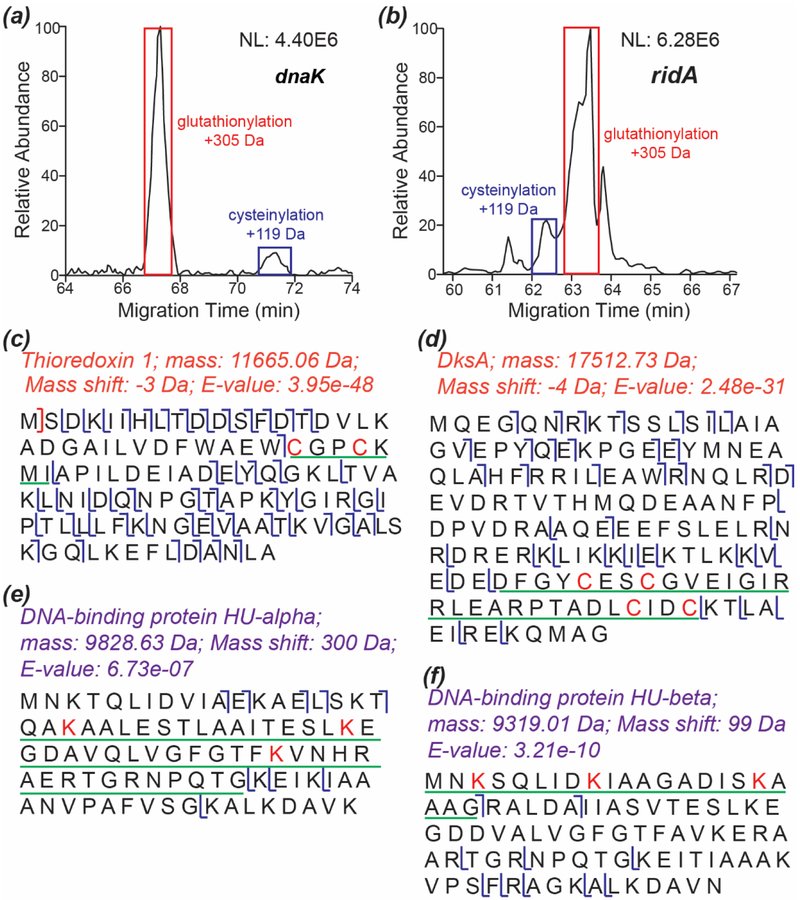
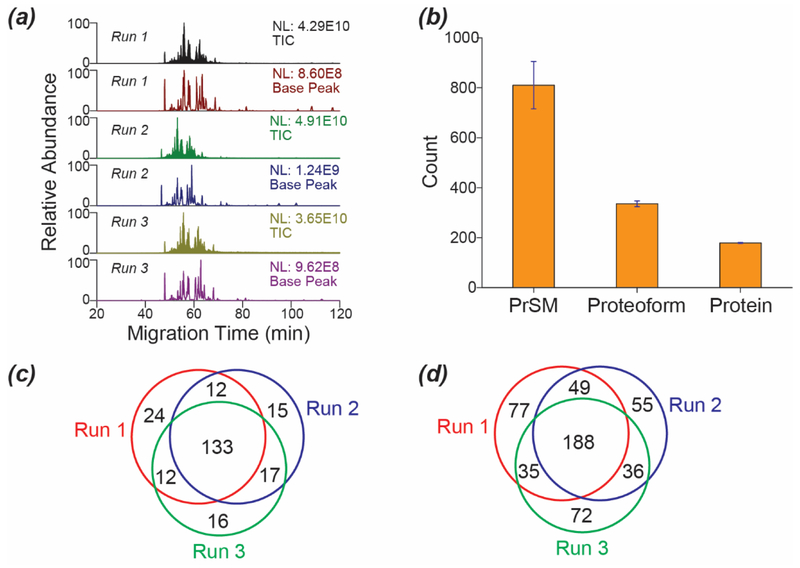
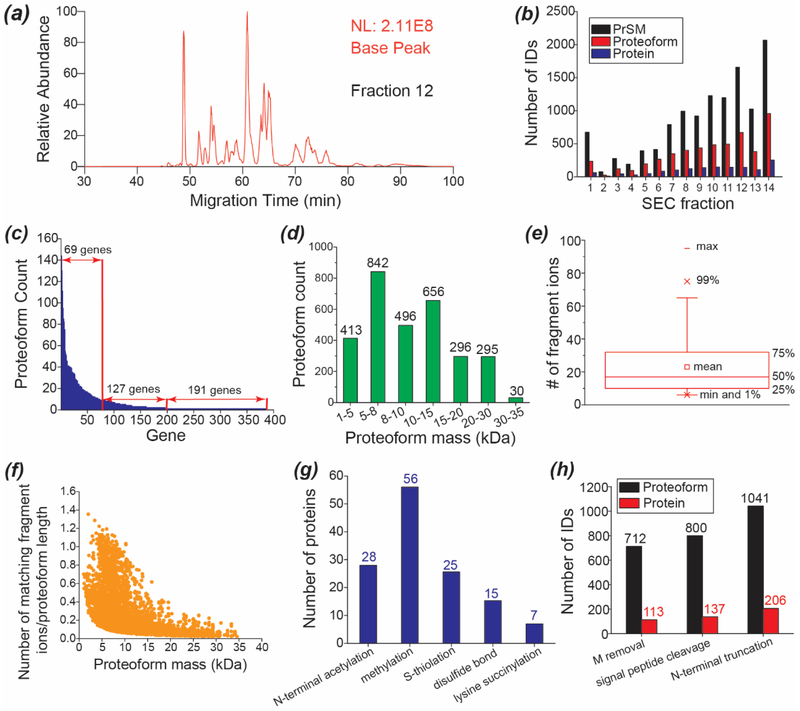
Capillary zone electrophoresis (CZE)-tandem mass spectrometry (MS/MS) has been recognized as an efficient approach for top-down proteomics recently for its high-capacity separation and highly sensitive detection of proteoforms. However, the commonly used collision-based dissociation methods often cannot provide extensive fragmentation of proteoforms for thorough characterization. Activated ion electron transfer dissociation (AI-ETD), that combines infrared photoactivation concurrent with ETD, has shown better performance for proteoform fragmentation than higher energy-collisional dissociation (HCD) and standard ETD. Here, we present the first application of CZE-AI-ETD on an Orbitrap Fusion Lumos mass spectrometer for large-scale top-down proteomics of Escherichia coli (E. coli) cells. CZE-AI-ETD outperformed CZE-ETD regarding proteoform and protein identifications (IDs). CZE-AI-ETD reached comparable proteoform and protein IDs with CZE-HCD. CZE-AI-ETD tended to generate better expectation values (E values) of proteoforms than CZE-HCD and CZE-ETD, indicating a higher quality of MS/MS spectra from AI-ETD respecting the number of sequence-informative fragment ions generated. CZE-AI-ETD showed great reproducibility regarding the proteoform and protein IDs with relative standard deviations less than 4% and 2% (n = 3). Coupling size exclusion chromatography (SEC) to CZE-AI-ETD identified 3028 proteoforms and 387 proteins from E. coli cells with 1% spectrum level and 5% proteoform-level false discovery rates. The data represents the largest top-down proteomics dataset using the AI-ETD method so far. Single-shot CZE-AI-ETD of one SEC fraction identified 957 proteoforms and 253 proteins. N-terminal truncations, signal peptide cleavage, N-terminal methionine removal, and various post-translational modifications including protein N-terminal acetylation, methylation, S-thiolation, disulfide bonds, and lysine succinylation were detected.
毛细管区带电泳(CZE)-串联质谱(MS/MS)最近因其对蛋白质异构体的高容量分离和高灵敏度检测而被公认为一种有效的自上而下蛋白质组学方法。然而,常用的基于碰撞的解离方法通常不能为彻底的特征描述提供广泛的蛋白质异构体片段。与更高能量碰撞解离(HCD)和标准 ETD 相比,结合红外光活化与 ETD 的活性离子电子转移解离(AI-ETD)在蛋白质异构体片段化方面表现出更好的性能。在这里,我们首次将 CZE-AI-ETD 应用于 Orbitrap Fusion Lumos 质谱仪,用于对大肠杆菌(E. coli)细胞进行大规模的自上而下蛋白质组学研究。CZE-AI-ETD 在蛋白质异构体和蛋白质鉴定(IDs)方面优于 CZE-ETD。CZE-AI-ETD 与 CZE-HCD 具有相当的蛋白质异构体和蛋白质 IDs。CZE-AI-ETD 生成的蛋白质异构体的预期值(E 值)往往优于 CZE-HCD 和 CZE-ETD,表明 AI-ETD 产生的 MS/MS 谱具有更高的质量,这与生成的序列信息片段离子数量有关。CZE-AI-ETD 在蛋白质异构体和蛋白质 IDs 方面具有很好的重现性,相对标准偏差小于 4%和 2%(n=3)。将尺寸排阻色谱(SEC)与 CZE-AI-ETD 相结合,从大肠杆菌细胞中鉴定出 3028 种蛋白质异构体和 387 种蛋白质,在谱水平上的假发现率为 1%,在蛋白质异构体水平上的假发现率为 5%。该数据集是迄今为止使用 AI-ETD 方法进行的最大规模的自上而下蛋白质组学数据集。对一个 SEC 馏分的单次 CZE-AI-ETD 鉴定出 957 种蛋白质异构体和 253 种蛋白质。检测到 N 端截短、信号肽切割、N 端甲硫氨酸切除以及各种翻译后修饰,包括蛋白质 N 端乙酰化、甲基化、S-硫代化、二硫键和赖氨酸琥珀酰化。