Indelicato Elisabetta, Boesch Sylvia
Center for Rare Movement Disorders Innsbruck, Department of Neurology, Medical University of Innsbruck, Innsbruck, Austria.
Front Neurol. 2021 Mar 2;12:639994. doi: 10.3389/fneur.2021.639994. eCollection 2021.
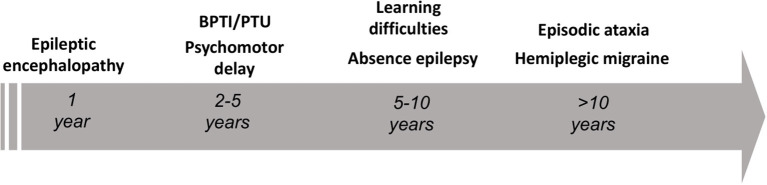
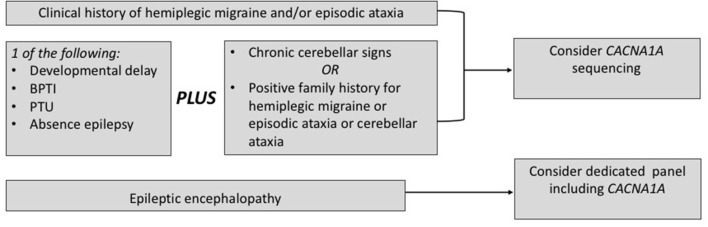
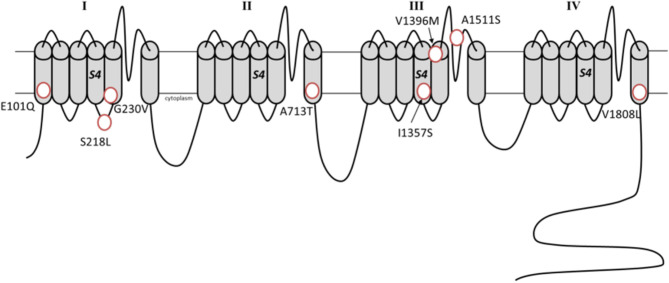
Ion channel dysfunction is a key pathological substrate of episodic neurological disorders. A classical gene associated to paroxysmal movement disorders is , which codes for the pore-forming subunit of the neuronal calcium channel P/Q. Non-polyglutamine variants underlie familial hemiplegic ataxia type 1 (FHM1) and episodic ataxia type 2 (EA2). Classical paroxysmal manifestations of FHM1 are migraine attacks preceded by motor aura consisting of hemiparesis, aphasia, and disturbances of consciousness until coma. Patients with EA2 suffer of recurrent episodes of vertigo, unbalance, diplopia, and vomiting. Beyond these typical presentations, several reports highlighted manifold clinical features associated with P/Q channelopathies, from chronic progressive cerebellar ataxia to epilepsy and psychiatric disturbances. These manifestations may often outlast the burden of classical episodic symptoms leading to pitfalls in the diagnostic work-up. Lately, the spreading of next generation sequencing techniques linked variants to an even broader phenotypic spectrum including early developmental delay, autism spectrum disorders, epileptic encephalopathy, and early onset paroxysmal dystonia. The age-dependency represents a striking new aspect of these phenotypes und highlights a pivotal role for P/Q channels in the development of the central nervous system in a defined time window. While several reviews addressed the clinical presentation and treatment of FHM1 and EA2, an overview of the newly described age-dependent manifestations is lacking. In this Mini-Review we present a clinical update, delineate genotype-phenotype correlations as well as summarize evidence on the pathophysiological mechanisms underlying the expanded phenotype associated with variants.
离子通道功能障碍是发作性神经系统疾病的关键病理基础。一个与阵发性运动障碍相关的经典基因是 ,它编码神经元钙通道P/Q的孔形成亚基。非多聚谷氨酰胺变异是1型家族性偏瘫性共济失调(FHM1)和2型发作性共济失调(EA2)的基础。FHM1的典型发作表现是偏头痛发作,之前有运动性先兆,包括偏瘫、失语和意识障碍直至昏迷。EA2患者会反复出现眩晕、失衡、复视和呕吐。除了这些典型表现外,一些报告强调了与P/Q通道病相关的多种临床特征,从慢性进行性小脑共济失调到癫痫和精神障碍。这些表现往往会持续超过经典发作症状的负担,导致诊断检查出现失误。最近,下一代测序技术的普及将 变异与更广泛的表型谱联系起来,包括早期发育迟缓、自闭症谱系障碍、癫痫性脑病和早发性阵发性肌张力障碍。年龄依赖性是这些表型的一个显著新特征,突出了P/Q通道在特定时间窗口中枢神经系统发育中的关键作用。虽然有几篇综述讨论了FHM1和EA2的临床表现和治疗,但缺乏对新描述的年龄依赖性表现的概述。在本综述中,我们介绍了临床最新情况,描述了基因型-表型相关性,并总结了与 变异相关的扩展表型的病理生理机制的证据。