Friedrich Baur Institute at the Department of Neurology, University Hospital, Ludwig-Maximilians-University Munich, 80336 Munich, Germany.
German Center for Neurodegenerative Diseases (DZNE), 81377 Munich, Germany.
Int J Mol Sci. 2020 May 27;21(11):3810. doi: 10.3390/ijms21113810.
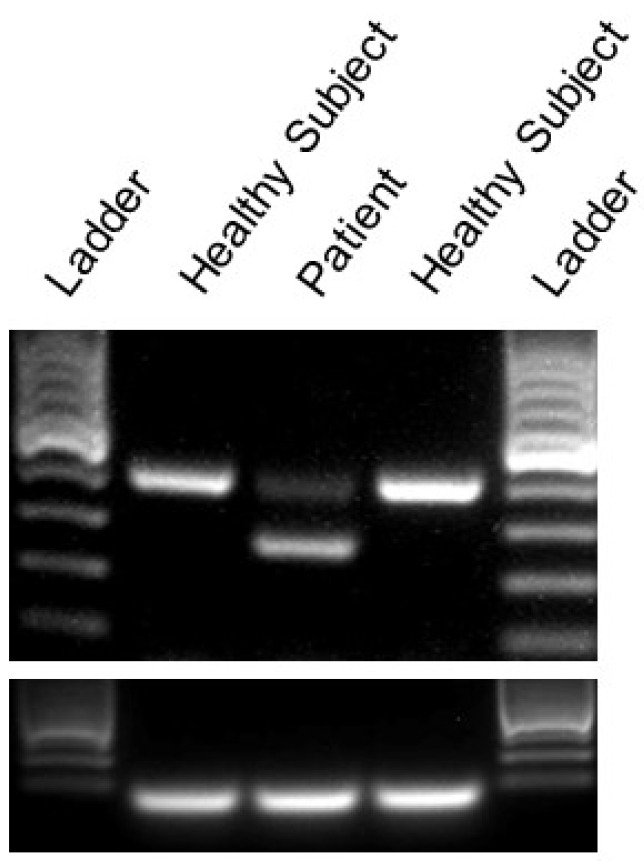
Episodic ataxia type 2 (EA2) is characterized by paroxysmal attacks of ataxia with typical onset in childhood or early adolescence. The disease is associated with mutations in the voltage-gated calcium channel alpha 1A subunit (Cav2.1) that is encoded by the gene. However, previously unrecognized atypical symptoms and the genetic overlap existing between EA2, spinocerebellar ataxia type 6, familial hemiplegic migraine type 1, and other neurological diseases blur the genotype/phenotype correlations, making a differential diagnosis difficult to formulate correctly and delaying early therapeutic intervention. Here we report a new clinical phenotype of a -associated disease characterized by absence epilepsy occurring during childhood. However, much later in life the patient displayed non-episodic, slowly progressive gait ataxia. Gene panel sequencing for hereditary ataxias led to the identification of a novel heterozygous mutation (c.1913 + 2T > G), altering the donor splice site of intron 14. This genetic defect was predicted to result in an in-frame deletion removing 44 amino acids from the voltage-gated calcium channel Cav2.1. An RT-PCR analysis of cDNA derived from patient skin fibroblasts confirmed the skipping of the entire exon 14. Furthermore, two-electrode voltage-clamp recordings performed from oocytes expressing a wild-type versus mutant channel showed that the genetic defect caused a complete loss of channel function. This represents the first description of distinct clinical manifestations that remarkably expand the genetic and phenotypic spectrum of related diseases and should be considered for an early diagnosis and effective therapeutic intervention.
发作性共济失调 2 型(EA2)的特征是阵发的共济失调发作,典型的发病年龄在儿童期或青春期早期。该疾病与电压门控钙通道 alpha 1A 亚基(Cav2.1)的突变有关,该基因由 基因编码。然而,以前未被认识到的非典型症状以及 EA2、脊髓小脑共济失调 6 型、家族性偏瘫性偏头痛 1 型和其他神经疾病之间的遗传重叠,模糊了基因型/表型相关性,使得正确制定鉴别诊断变得困难,并延迟了早期的治疗干预。在这里,我们报告了一种新的与 - 相关疾病的临床表型,其特征是儿童时期发生的失神发作。然而,在生命的后期,患者表现出非阵发性、缓慢进展的步态共济失调。遗传性共济失调的基因面板测序导致发现了一种新的杂合 突变(c.1913 + 2T > G),改变了内含子 14 的供体位点。这种遗传缺陷预计会导致一个框内缺失,从电压门控钙通道 Cav2.1 中删除 44 个氨基酸。从患者皮肤成纤维细胞中提取的 cDNA 的 RT-PCR 分析证实了整个外显子 14 的跳过。此外,在表达野生型与突变通道的卵母细胞中进行的双电极电压钳记录表明,遗传缺陷导致通道功能完全丧失。这是首次描述明显扩展相关疾病的遗传和表型谱的不同临床表现,应考虑早期诊断和有效治疗干预。