Qian Xinye, Wang Jun, Wang Meng, Igelman Austin D, Jones Kaylie D, Li Yumei, Wang Keqing, Goetz Kerry E, Birch David G, Yang Paul, Pennesi Mark E, Chen Rui
Verna and Marrs McLean Department of Biochemistry and Molecular Biology, Baylor College of Medicine, Houston, TX, United States.
Human Genome Sequencing Center, Baylor College of Medicine, Houston, TX, United States.
Front Genet. 2021 Mar 2;12:647400. doi: 10.3389/fgene.2021.647400. eCollection 2021.
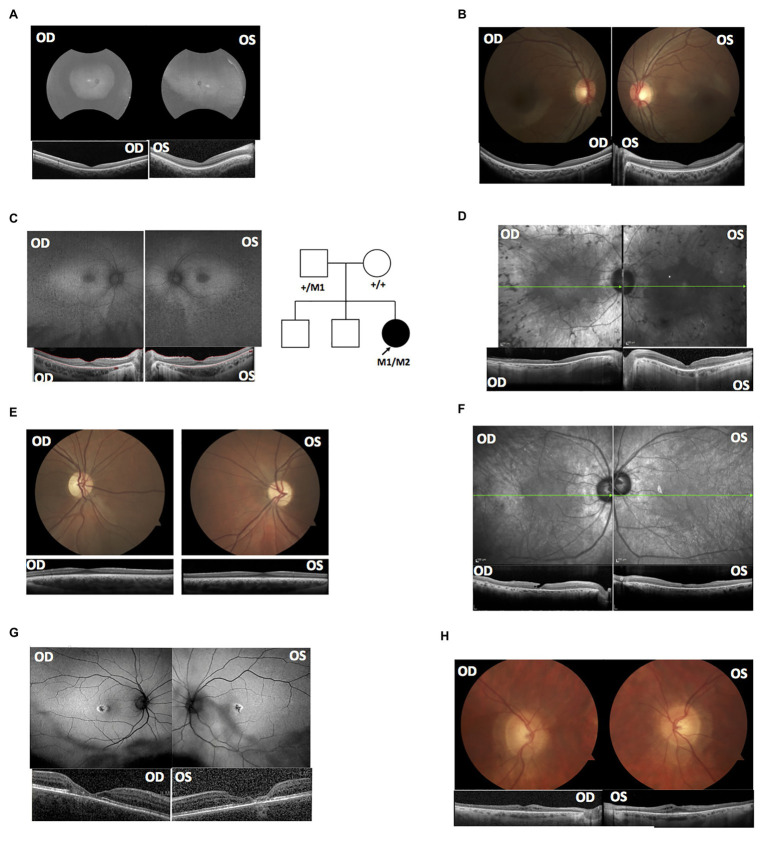
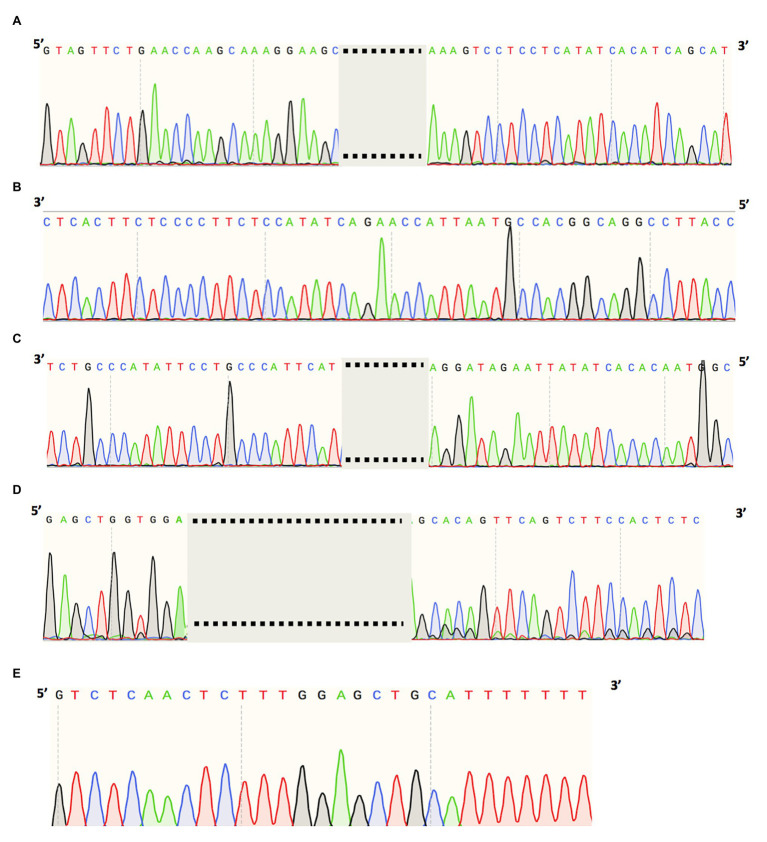
High throughput sequencing technologies have revolutionized the identification of mutations responsible for a diverse set of Mendelian disorders, including inherited retinal disorders (IRDs). However, the causal mutations remain elusive for a significant proportion of patients. This may be partially due to pathogenic mutations located in non-coding regions, which are largely missed by capture sequencing targeting the coding regions. The advent of whole-genome sequencing (WGS) allows us to systematically detect non-coding variations. However, the interpretation of these variations remains a significant bottleneck. In this study, we investigated the contribution of deep-intronic splice variants to IRDs. WGS was performed for a cohort of 571 IRD patients who lack a confident molecular diagnosis, and potential deep intronic variants that affect proper splicing were identified using SpliceAI. A total of six deleterious deep intronic variants were identified in eight patients. An minigene system was applied to further validate the effect of these variants on the splicing pattern of the associated genes. The prediction scores assigned to splice-site disruption positively correlated with the impact of mutations on splicing, as those with lower prediction scores demonstrated partial splicing. Through this study, we estimated the contribution of deep-intronic splice mutations to unassigned IRD patients and leveraged and methods to establish a framework for prioritizing deep intronic variant candidates for mechanistic and functional analyses.
高通量测序技术彻底改变了对导致多种孟德尔疾病(包括遗传性视网膜疾病,IRD)的突变的识别。然而,对于相当一部分患者来说,致病突变仍然难以捉摸。这可能部分归因于位于非编码区的致病突变,而针对编码区的捕获测序很大程度上会遗漏这些突变。全基因组测序(WGS)的出现使我们能够系统地检测非编码变异。然而,对这些变异的解读仍然是一个重大瓶颈。在本研究中,我们调查了深度内含子剪接变异对IRD的影响。对571名缺乏可靠分子诊断的IRD患者进行了WGS,并使用SpliceAI识别了可能影响正确剪接的潜在深度内含子变异。在8名患者中总共鉴定出6个有害的深度内含子变异。应用一个小型基因系统进一步验证这些变异对相关基因剪接模式的影响。分配给剪接位点破坏的预测分数与突变对剪接的影响呈正相关,因为预测分数较低的变异表现出部分剪接。通过这项研究,我们估计了深度内含子剪接突变对未确诊IRD患者的影响,并利用相关方法建立了一个框架,用于对深度内含子变异候选物进行优先排序,以便进行机制和功能分析。