Medicine Genetics Group, Vall d'Hebron Research Institute (VHIR), Barcelona, Spain.
Department of Clinical and Molecular Genetics, Hospital Vall d'Hebron, Barcelona, Spain.
Hum Mutat. 2021 Jun;42(6):787-795. doi: 10.1002/humu.24200. Epub 2021 Apr 6.
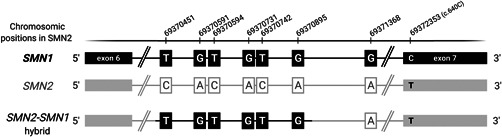
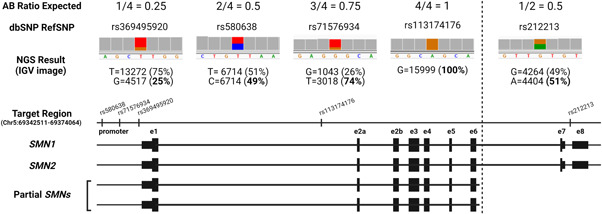
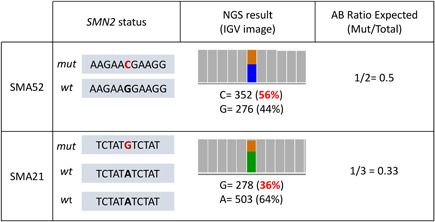
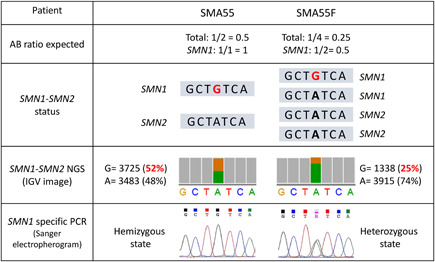
Spinal muscular atrophy (SMA) is caused by bi-allelic loss or pathogenic variants in the SMN1 gene. SMN2, the highly homologous copy of SMN1, is considered the major phenotypic modifier of the disease. Determination of SMN2 copy number is essential to establish robust genotype-phenotype correlations and predict disease evolution, to stratify patients for clinical trials, as well as to define those eligible for treatment. Discordant genotype-phenotype correlations are not uncommon in SMA, some of which are due to intragenic SMN2 variants that may influence the amount of complete SMN transcripts and, therefore, of full-length SMN protein. Detection of these variants is crucial to predict SMA phenotypes in the present scenario of therapeutic advances and with the perspective of SMA neonatal screening and early diagnosis to start treatments. Here, we present a novel, affordable, and versatile method for complete sequencing of the SMN2 gene based on long-range polymerase chain reaction and next-generation sequencing. The method was validated by analyzing samples from 53 SMA patients who lack SMN1, allowing to characterize paralogous, rare variants, and single-nucleotide polymorphisms of SMN2 as well as SMN2-SMN1 hybrid genes. The method identifies partial deletions and can be adapted to determine rare pathogenic variants in patients with at least one SMN1 copy.
脊髓性肌萎缩症(SMA)是由 SMN1 基因的双等位基因缺失或致病变异引起的。SMN2 是 SMN1 的高度同源拷贝,被认为是疾病的主要表型修饰因子。确定 SMN2 拷贝数对于建立稳健的基因型-表型相关性和预测疾病演变、对临床试验进行分层以及定义那些有资格接受治疗的患者至关重要。SMA 中并不罕见存在不一致的基因型-表型相关性,其中一些是由于基因内 SMN2 变异,这些变异可能影响完整 SMN 转录本的数量,从而影响全长 SMN 蛋白的数量。在治疗进展的当前情况下,以及从 SMA 新生儿筛查和早期诊断的角度出发,为了开始治疗,检测这些变异至关重要。在这里,我们提出了一种基于长距离聚合酶链反应和下一代测序的新颖、经济实惠且多功能的 SMN2 基因完整测序方法。该方法通过分析 53 名缺乏 SMN1 的 SMA 患者的样本进行了验证,能够对 SMN2 的等位基因、罕见变异和单核苷酸多态性以及 SMN2-SMN1 杂交基因进行特征描述。该方法可识别部分缺失,并可适用于确定至少有一个 SMN1 拷贝的患者中的罕见致病性变异。