Zwartkruis M M, Elferink M G, Gommers D, Signoria I, Blasco-Pérez L, Costa-Roger M, van der Sel J, Renkens I J, Green J W, Kortooms J V, Vermeulen C, Straver R, van Deutekom H W M, Veldink J H, Asselman F, Tizzano E F, Wadman R I, van der Pol W L, van Haaften G W, Groen E J N
Department of Neurology and Neurosurgery, UMC Utrecht Brain Center, University Medical Center Utrecht, Utrecht, the Netherlands.
Department of Genetics, University Medical Center Utrecht, Utrecht, the Netherlands.
Genome Med. 2025 Mar 21;17(1):26. doi: 10.1186/s13073-025-01448-2.
The complex 2 Mb survival motor neuron (SMN) locus on chromosome 5q13, including the spinal muscular atrophy (SMA)-causing gene SMN1 and modifier SMN2, remains incompletely resolved due to numerous segmental duplications. Variation in SMN2 copy number, presumably influenced by SMN1 to SMN2 gene conversion, affects disease severity, though SMN2 copy number alone has insufficient prognostic value due to limited genotype-phenotype correlations. With advancements in newborn screening and SMN-targeted therapies, identifying genetic markers to predict disease progression and treatment response is crucial. Progress has thus far been limited by methodological constraints.
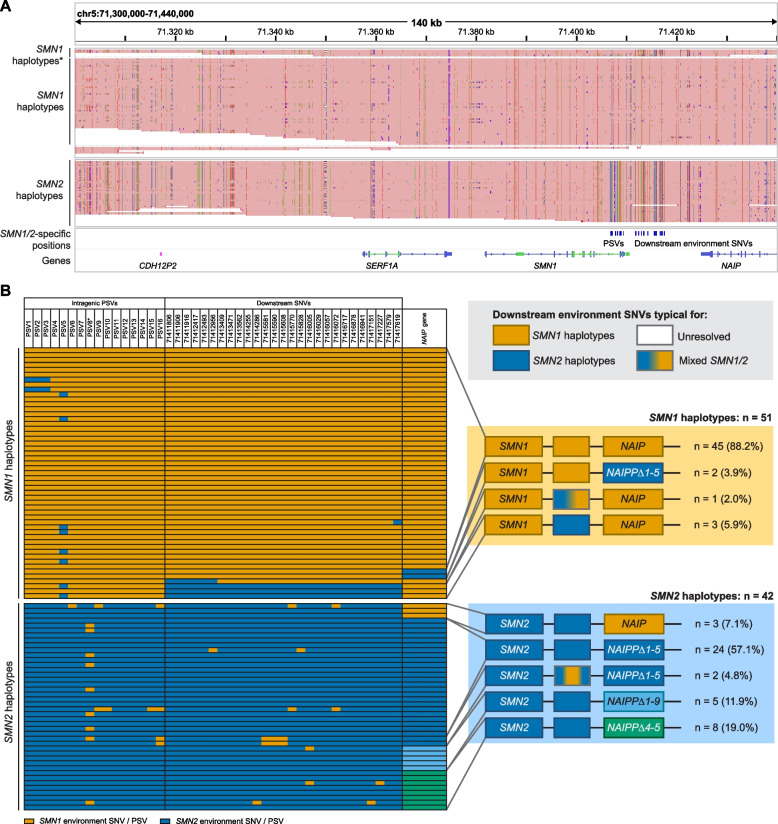
To address this, we developed HapSMA, a method to perform polyploid phasing of the SMN locus to enable copy-specific analysis of SMN and its surrounding genes. We used HapSMA on publicly available Oxford Nanopore Technologies (ONT) sequencing data of 29 healthy controls and performed long-read, targeted ONT sequencing of the SMN locus of 31 patients with SMA.
In healthy controls, we identified single nucleotide variants (SNVs) specific to SMN1 and SMN2 haplotypes that could serve as gene conversion markers. Broad phasing including the NAIP gene allowed for a more complete view of SMN locus variation. Genetic variation in SMN2 haplotypes was larger in SMA patients. Forty-two percent of SMN2 haplotypes of SMA patients showed varying SMN1 to SMN2 gene conversion breakpoints, serving as direct evidence of gene conversion as a common genetic characteristic in SMA and highlighting the importance of inclusion of SMA patients when investigating the SMN locus.
Our findings illustrate that both methodological advances and the analysis of patient samples are required to advance our understanding of complex genetic loci and address critical clinical challenges.
位于5号染色体q13区域的复杂的2兆碱基生存运动神经元(SMN)基因座,包括导致脊髓性肌萎缩症(SMA)的基因SMN1和修饰基因SMN2,由于存在大量节段性重复,其结构仍未完全解析。SMN2拷贝数的变化可能受SMN1向SMN2基因转换的影响,进而影响疾病严重程度,不过由于基因型与表型的相关性有限,仅SMN2拷贝数的预后价值不足。随着新生儿筛查和针对SMN的治疗方法的进展,识别预测疾病进展和治疗反应的遗传标记至关重要。然而,迄今为止,进展因方法学限制而受限。
为解决这一问题,我们开发了HapSMA方法,用于对SMN基因座进行多倍体定相,以实现对SMN及其周围基因的拷贝特异性分析。我们将HapSMA应用于29名健康对照者公开可用的牛津纳米孔技术(ONT)测序数据,并对31例SMA患者的SMN基因座进行了长读长靶向ONT测序。
在健康对照者中,我们鉴定出了特定于SMN1和SMN2单倍型的单核苷酸变异(SNV),可作为基因转换标记。包括NAIP基因在内的广泛定相使我们能够更全面地了解SMN基因座变异。SMA患者中SMN2单倍型的遗传变异更大。42%的SMA患者的SMN2单倍型显示出不同程度的SMN1向SMN2基因转换断点,这直接证明了基因转换是SMA的一种常见遗传特征,并突出了在研究SMN基因座时纳入SMA患者的重要性。
我们的研究结果表明,要增进对复杂基因座的理解并应对关键临床挑战,既需要方法学上的进步,也需要对患者样本进行分析。