Department of Molecular Medicine, The Scripps Research Institute, La Jolla, CA 92037.
School of Public Health (Shenzhen), Sun Yat-sen University, 510006 Guangzhou, China.
Proc Natl Acad Sci U S A. 2021 Mar 30;118(13). doi: 10.1073/pnas.2012898118.
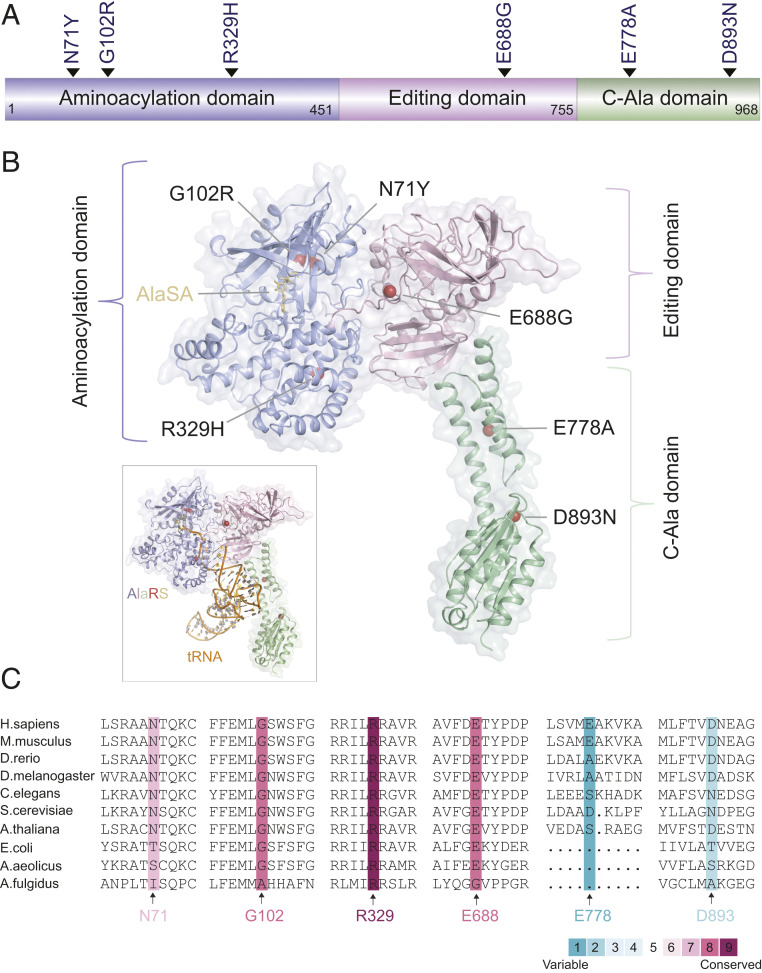
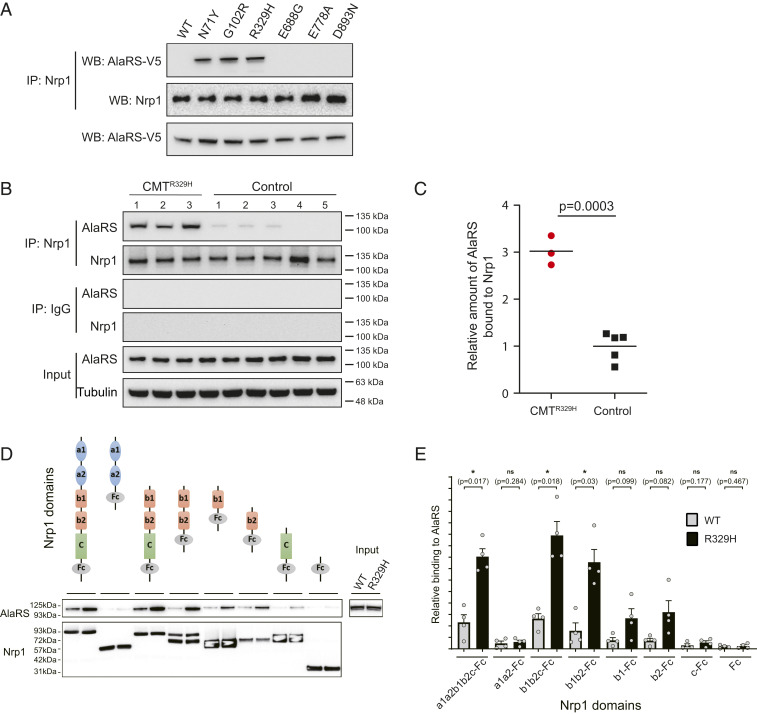
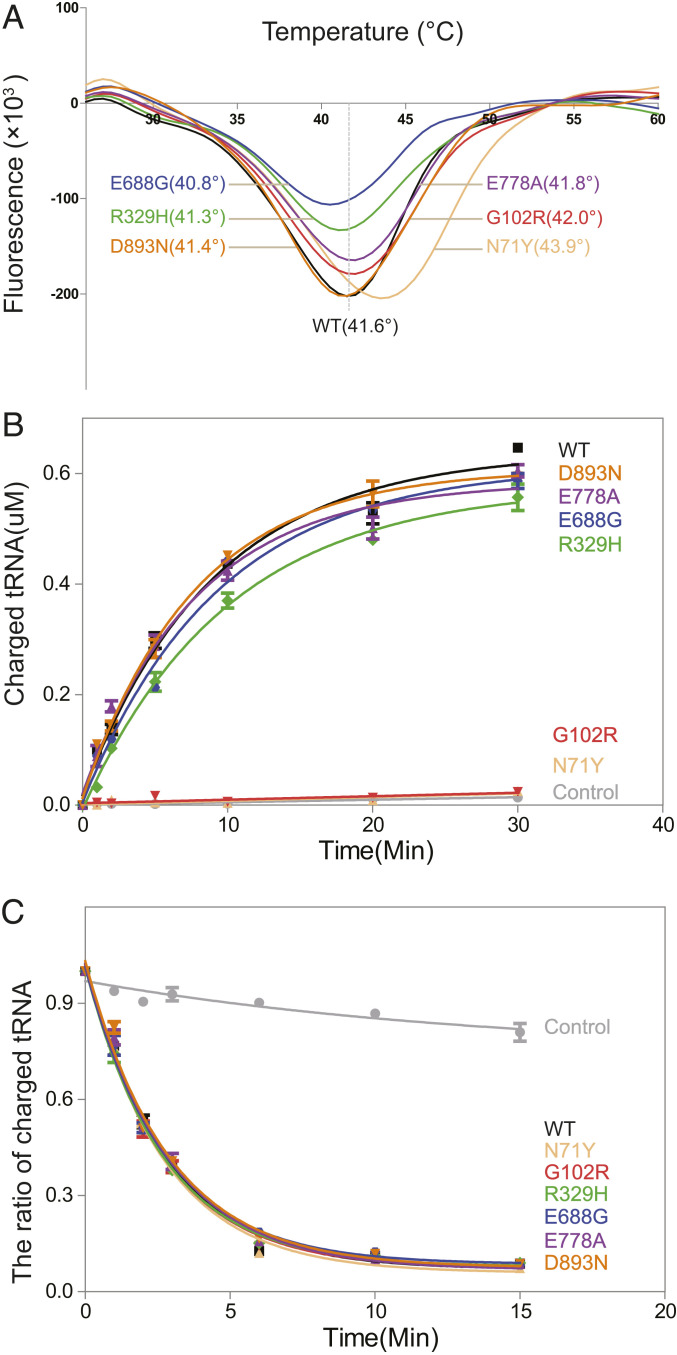
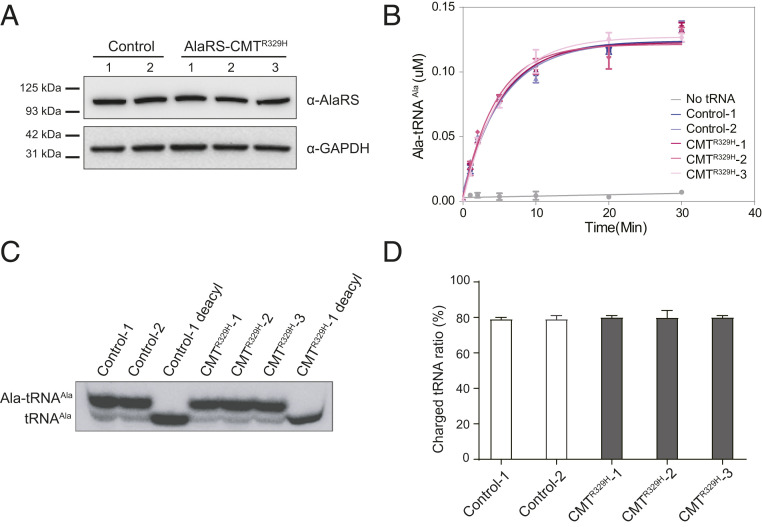
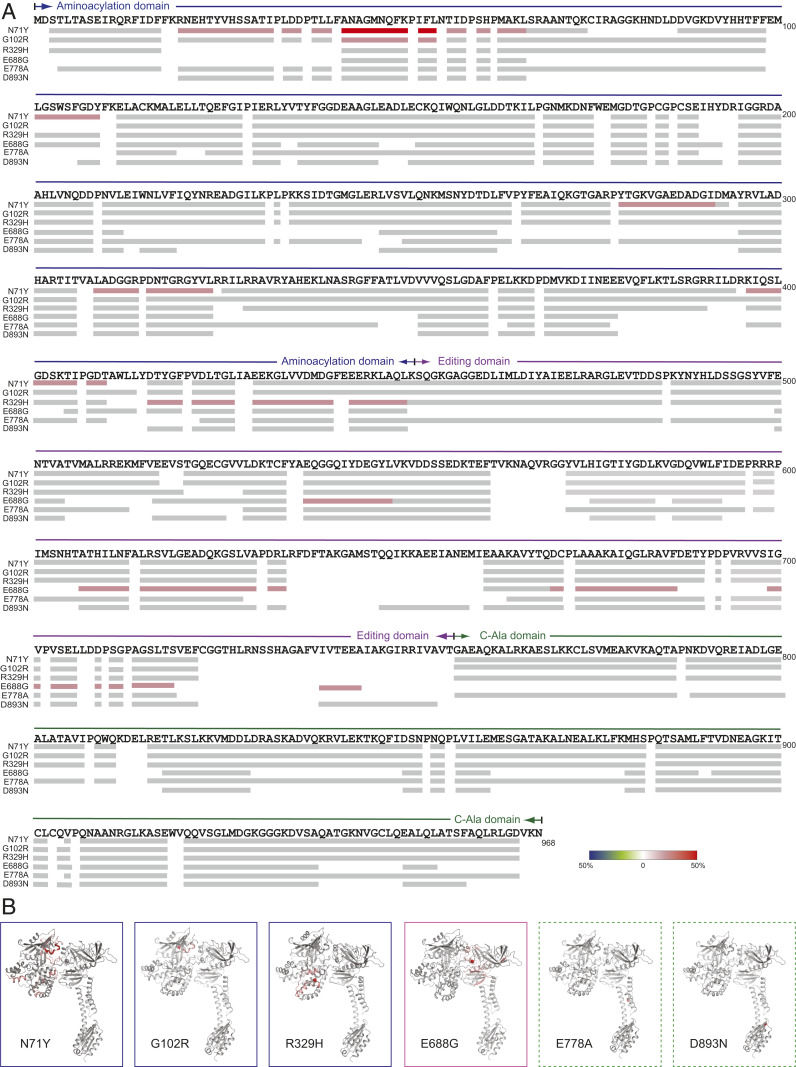
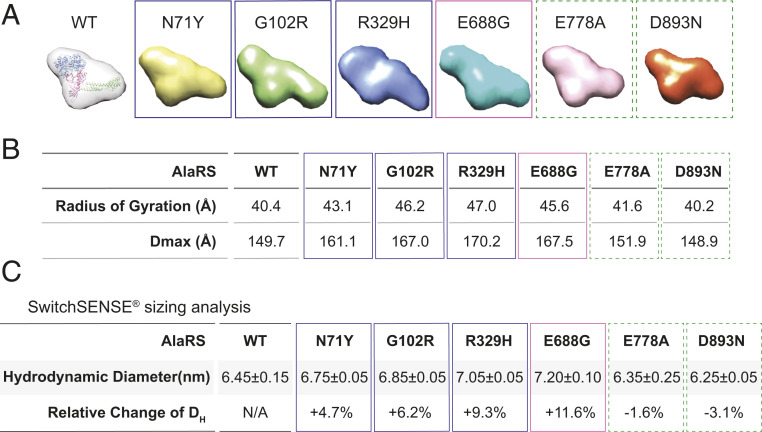
Through dominant mutations, aminoacyl-tRNA synthetases constitute the largest protein family linked to Charcot-Marie-Tooth disease (CMT). An example is CMT subtype 2N (CMT2N), caused by individual mutations spread out in AlaRS, including three in the aminoacylation domain, thereby suggesting a role for a tRNA-charging defect. However, here we found that two are aminoacylation defective but that the most widely distributed R329H is normal as a purified protein in vitro and in unfractionated patient cell samples. Remarkably, in contrast to wild-type (WT) AlaRS, all three mutant proteins gained the ability to interact with neuropilin 1 (Nrp1), the receptor previously linked to CMT pathogenesis in GlyRS. The aberrant AlaRS-Nrp1 interaction is further confirmed in patient samples carrying the R329H mutation. However, CMT2N mutations outside the aminoacylation domain do not induce the Nrp1 interaction. Detailed biochemical and biophysical investigations, including X-ray crystallography, small-angle X-ray scattering, hydrogen-deuterium exchange (HDX), switchSENSE hydrodynamic diameter determinations, and protease digestions reveal a mutation-induced structural loosening of the aminoacylation domain that correlates with the Nrp1 interaction. The b1b2 domains of Nrp1 are responsible for the interaction with R329H AlaRS. The results suggest Nrp1 is more broadly associated with CMT-associated members of the tRNA synthetase family. Moreover, we revealed a distinct structural loosening effect induced by a mutation in the editing domain and a lack of conformational impact with C-Ala domain mutations, indicating mutations in the same protein may cause neuropathy through different mechanisms. Our results show that, as with other CMT-associated tRNA synthetases, aminoacylation per se is not relevant to the pathology.
通过显性突变,氨酰-tRNA 合成酶构成了与遗传性运动感觉神经病(CMT)相关的最大蛋白质家族。CMT 亚型 2N(CMT2N)就是一个例子,它是由丙氨酰-tRNA 合成酶(AlaRS)中散布的个体突变引起的,包括三个在氨酰化结构域的突变,这表明 tRNA 酰基化缺陷可能与疾病相关。然而,我们发现其中两个突变体是氨酰化缺陷的,但分布最广的 R329H 在体外和未经分离的患者细胞样本中作为纯化蛋白是正常的。值得注意的是,与野生型(WT)AlaRS 相反,所有三种突变蛋白都获得了与神经纤毛蛋白 1(Nrp1)相互作用的能力,Nrp1 是先前与 GlyRS 相关的 CMT 发病机制的受体。在携带 R329H 突变的患者样本中进一步证实了异常的 AlaRS-Nrp1 相互作用。然而,氨酰化结构域外的 CMT2N 突变不会诱导 Nrp1 相互作用。详细的生化和生物物理研究,包括 X 射线晶体学、小角度 X 射线散射、氢氘交换(HDX)、switchSENSE 水动力直径测定和蛋白酶消化,揭示了突变诱导的氨酰化结构域结构松弛,与 Nrp1 相互作用相关。Nrp1 的 b1b2 结构域负责与 R329H AlaRS 相互作用。结果表明,Nrp1 与 tRNA 合成酶家族中与 CMT 相关的成员更广泛相关。此外,我们揭示了编辑结构域突变引起的明显结构松弛效应,以及 C-Ala 结构域突变缺乏构象影响,表明同一蛋白的突变可能通过不同的机制引起神经病。我们的结果表明,与其他与 CMT 相关的 tRNA 合成酶一样,氨酰化本身与病理学无关。