Portela Susana, Cabrera-Trujillo Jorge J, Fernández Israel
Departamento de Química Orgánica I and Centro de Innovación en Química Avanzada (ORFEO-CINQA), Facultad de Ciencias Químicas, Universidad Complutense de Madrid, 28040 Madrid, Spain.
J Org Chem. 2021 Apr 2;86(7):5317-5326. doi: 10.1021/acs.joc.1c00534. Epub 2021 Mar 25.
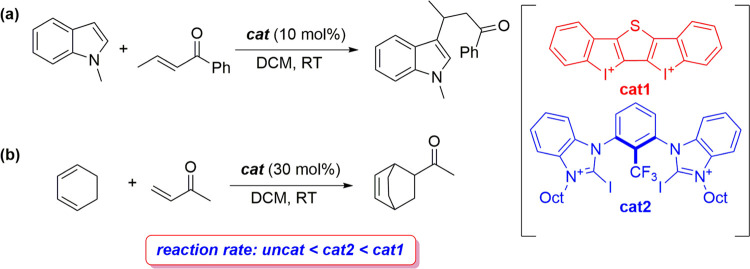
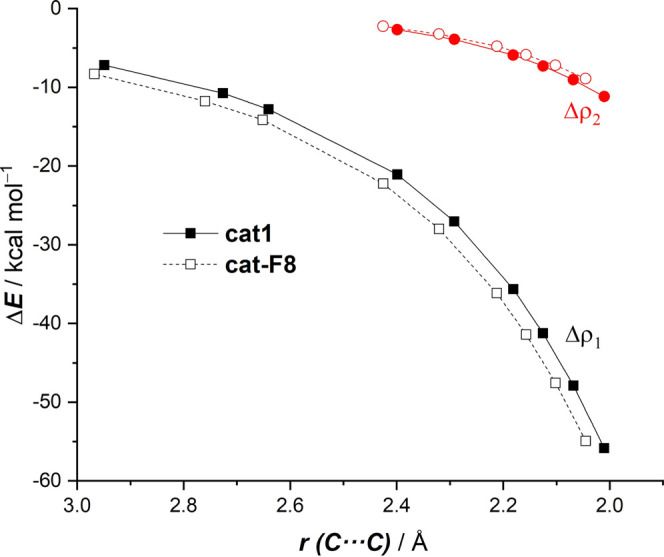
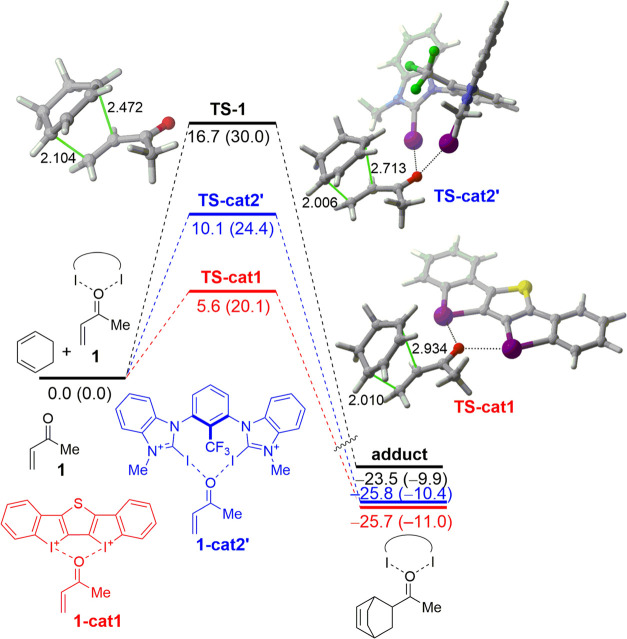
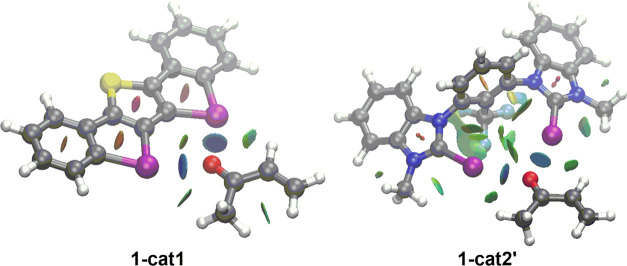
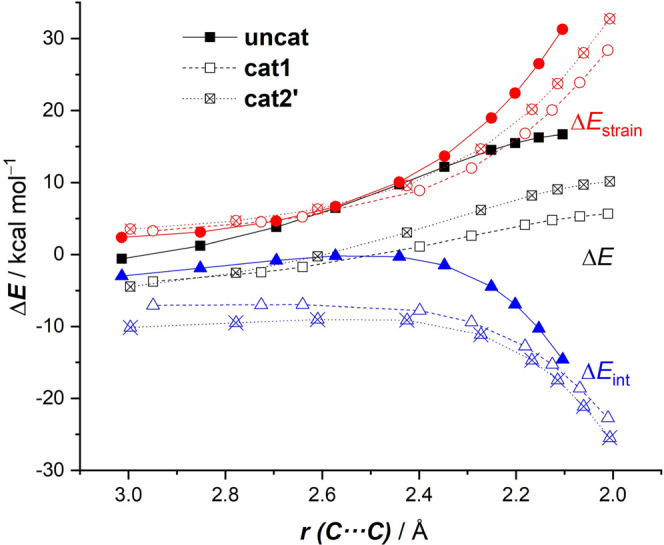
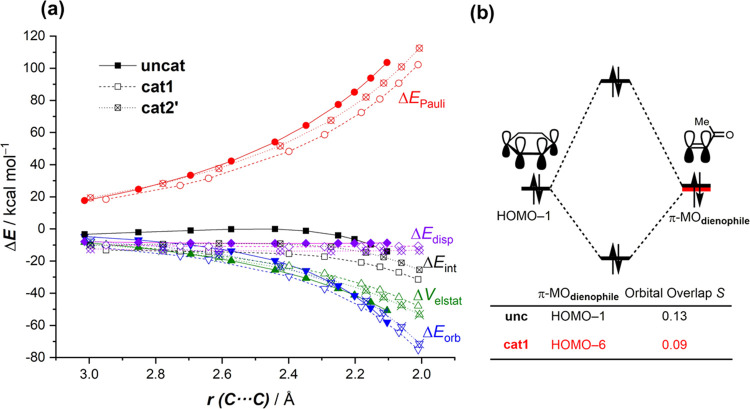
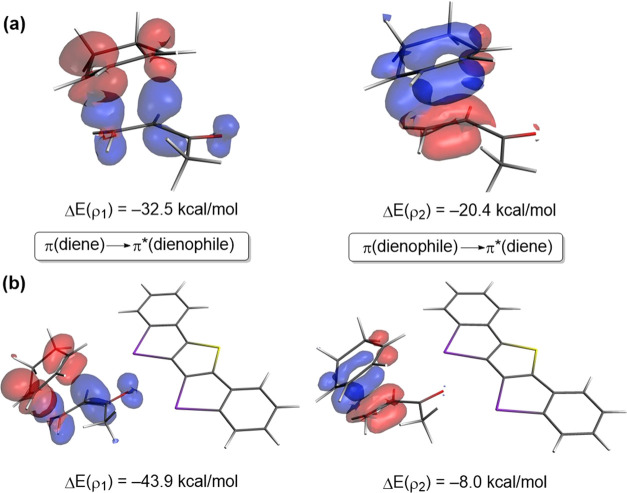
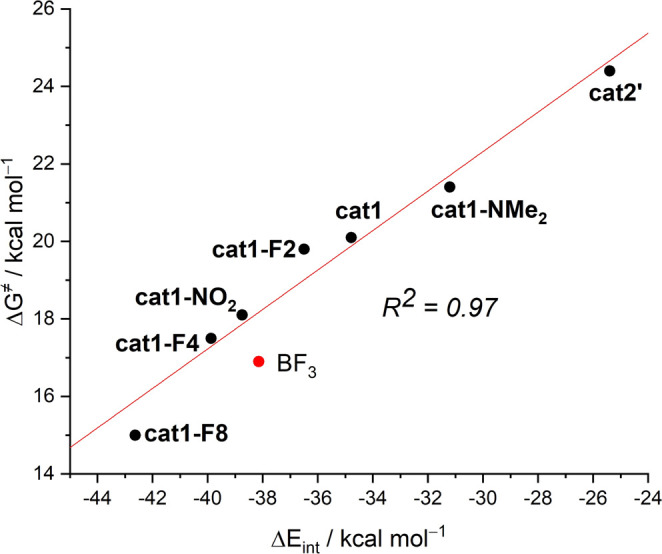
The poorly understood mode of activation and catalysis of bidentate iodine(III)-based halogen donors have been quantitatively explored in detail by means of state-of-the-art computational methods. To this end, the uncatalyzed Diels-Alder cycloaddition reaction between cyclohexadiene and methyl vinyl ketone is compared to the analogous process mediated by a bidentate iodine(III)-organocatalyst and by related, highly active iodine(I) species. It is found that the bidentate iodine(III)-catalyst accelerates the cycloaddition by lowering the reaction barrier up to 10 kcal mol compared to the parent uncatalyzed reaction. Our quantitative analyses reveal that the origin of the catalysis is found in a significant reduction of the steric (Pauli) repulsion between the diene and dienophile, which originates from both a more asynchronous reaction mode and a significant polarization of the π-system of the dienophile away from the incoming diene. Notably, the activity of the iodine(III)-catalyst can be further enhanced by increasing the electrophilic nature of the system. Thus, novel systems are designed whose activity actually surpasses that of strong Lewis acids such as BF.
利用最先进的计算方法,对基于双齿碘(III)的卤素供体的活化和催化模式进行了详细的定量研究,尽管人们对其了解甚少。为此,将环己二烯与甲基乙烯基酮之间的无催化狄尔斯-阿尔德环加成反应与由双齿碘(III)有机催化剂和相关的高活性碘(I)物种介导的类似过程进行了比较。研究发现,与未催化的原始反应相比,双齿碘(III)催化剂通过将反应势垒降低高达10千卡/摩尔来加速环加成反应。我们的定量分析表明,催化作用的起源在于二烯和亲双烯体之间空间(泡利)排斥的显著降低,这源于更不同步的反应模式以及亲双烯体π体系远离进入的二烯的显著极化。值得注意的是,通过增加体系的亲电性质,可以进一步提高碘(III)催化剂的活性。因此,设计了新型体系,其活性实际上超过了诸如BF等强路易斯酸的活性。