Institute for Diabetes Research and Metabolic Diseases of the Helmholtz Center Munich at the Eberhard Karls University of Tuebingen (IDM), Tuebingen, Germany.
Internal Medicine IV, Endocrinology, Diabetology and Nephrology, University Hospital Tuebingen, Tuebingen, Germany.
Diabetologia. 2021 Jun;64(6):1358-1374. doi: 10.1007/s00125-021-05435-1. Epub 2021 Mar 25.
AIMS/HYPOTHESIS: Neonatal beta cells carry out a programme of postnatal functional maturation to achieve full glucose responsiveness. A partial loss of the mature phenotype of adult beta cells may contribute to a reduction of functional beta cell mass and accelerate the onset of type 2 diabetes. We previously found that fetuin-A, a hepatokine increasingly secreted by the fatty liver and a determinant of type 2 diabetes, inhibits glucose-stimulated insulin secretion (GSIS) of human islets. Since fetuin-A is a ubiquitous fetal glycoprotein that declines peripartum, we examined here whether fetuin-A interferes with the functional maturity of beta cells.
The effects of fetuin-A were assessed during in vitro maturation of porcine neonatal islet cell clusters (NICCs) and in adult human islets. Expression alterations were examined via microarray, RNA sequencing and reverse transcription quantitative real-time PCR (qRT-PCR), proteins were analysed by western blotting and immunostaining, and insulin secretion was quantified in static incubations.
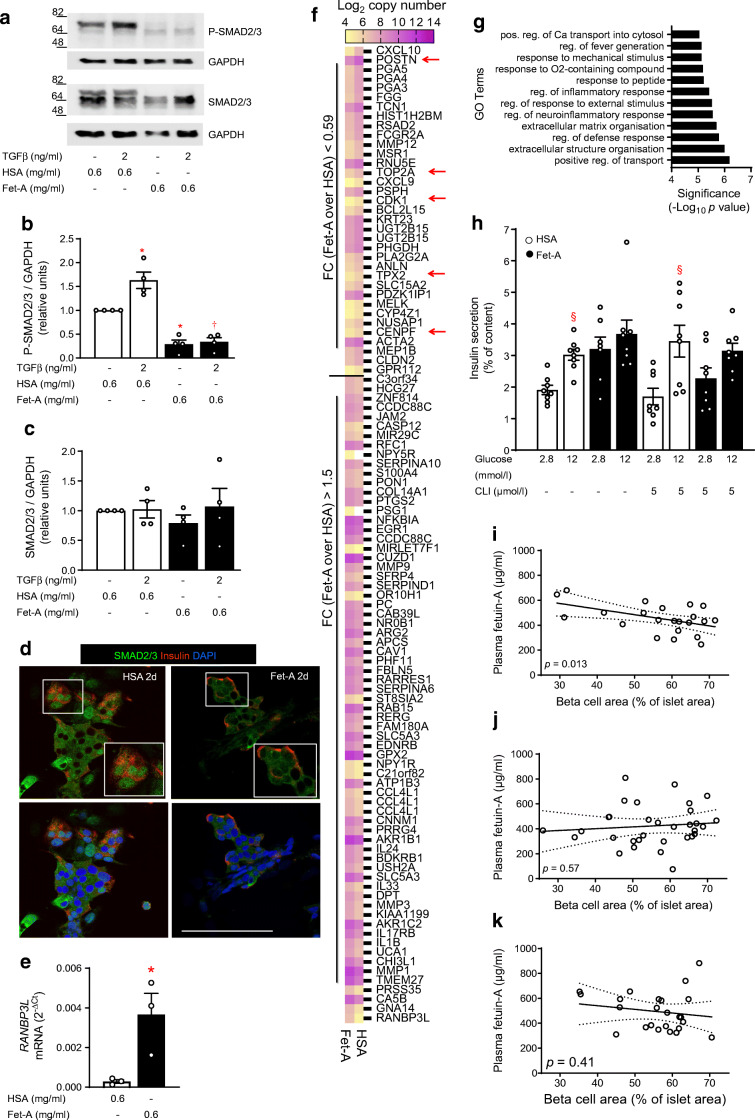
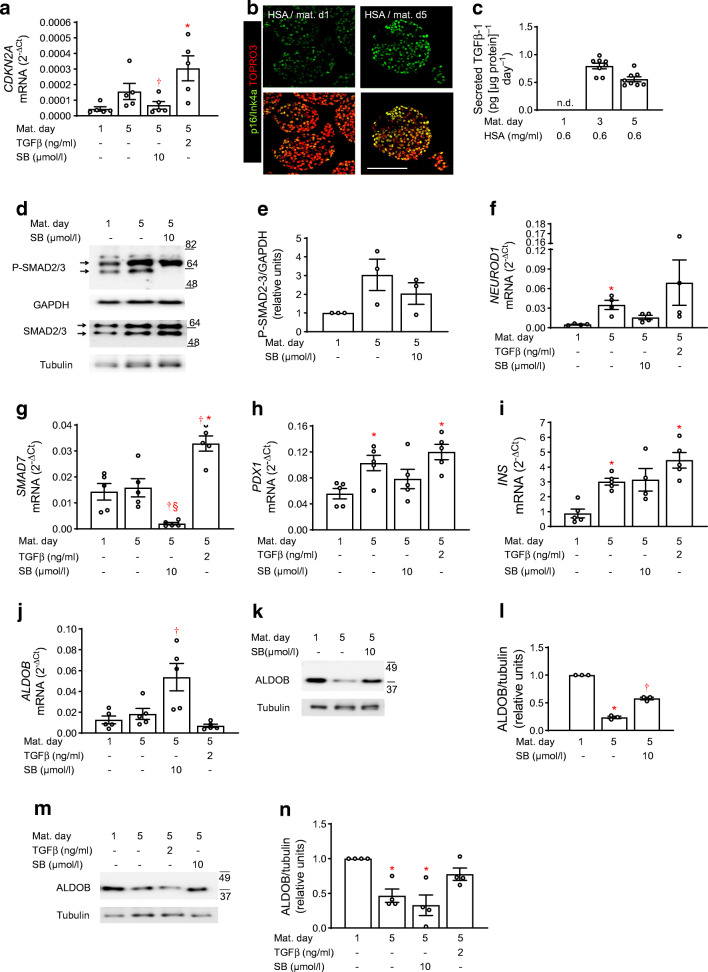
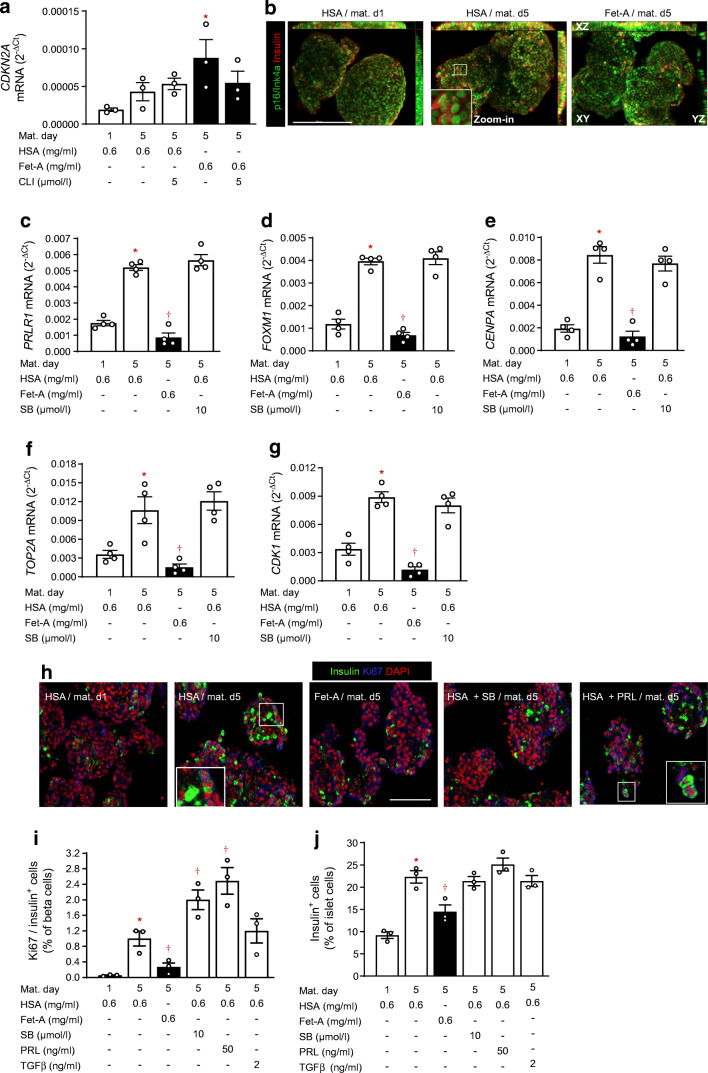
NICC maturation was accompanied by the gain of glucose-responsive insulin secretion (twofold stimulation), backed up by mRNA upregulation of genes governing beta cell identity and function, such as NEUROD1, UCN3, ABCC8 and CASR (Log fold change [LogFC] > 1.6). An active TGFβ receptor (TGFBR)-SMAD2/3 pathway facilitates NICC maturation, since the TGFBR inhibitor SB431542 counteracted the upregulation of aforementioned genes and de-repressed ALDOB, a gene disallowed in mature beta cells. In fetuin-A-treated NICCs, upregulation of beta cell markers and the onset of glucose responsiveness were suppressed. Concomitantly, SMAD2/3 phosphorylation was inhibited. Transcriptome analysis confirmed inhibitory effects of fetuin-A and SB431542 on TGFβ-1- and SMAD2/3-regulated transcription. However, contrary to SB431542 and regardless of cMYC upregulation, fetuin-A inhibited beta cell proliferation (0.27 ± 0.08% vs 1.0 ± 0.1% Ki67-positive cells in control NICCs). This effect was sustained by reduced expression (LogFC ≤ -2.4) of FOXM1, CENPA, CDK1 or TOP2A. In agreement, the number of insulin-positive cells was lower in fetuin-A-treated NICCs than in control NICCs (14.4 ± 1.2% and 22.3 ± 1.1%, respectively). In adult human islets fetuin-A abolished glucose responsiveness, i.e. 1.7- and 1.1-fold change over 2.8 mmol/l glucose in control- and fetuin-A-cultured islets, respectively. In addition, fetuin-A reduced SMAD2/3 phosphorylation and suppressed expression of proliferative genes. Of note, in non-diabetic humans, plasma fetuin-A was negatively correlated (p = 0.013) with islet beta cell area.
CONCLUSIONS/INTERPRETATION: Our results suggest that the perinatal decline of fetuin-A relieves TGFBR signalling in islets, a process that facilitates functional maturation of neonatal beta cells. Functional maturity remains revocable in later life, and the occurrence of a metabolically unhealthy milieu, such as liver steatosis and elevated plasma fetuin-A, can impair both function and adaptive proliferation of beta cells.
The RNAseq datasets and computer code produced in this study are available in the Gene Expression Omnibus (GEO): GSE144950; https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE144950.
目的/假设:新生儿β细胞进行出生后功能成熟的程序,以实现完全的葡萄糖反应性。成年β细胞成熟表型的部分丧失可能导致功能性β细胞质量的减少,并加速 2 型糖尿病的发生。我们之前发现,胎球蛋白-A(一种越来越多地由脂肪肝分泌的肝源激素,也是 2 型糖尿病的决定因素)抑制人胰岛的葡萄糖刺激胰岛素分泌(GSIS)。由于胎球蛋白-A 是一种普遍存在的胎儿糖蛋白,在围产期下降,我们在这里检查胎球蛋白-A 是否干扰β细胞的功能成熟。
在猪新生胰岛细胞簇(NICCs)的体外成熟过程中和在成人胰岛中评估胎球蛋白-A 的作用。通过微阵列、RNA 测序和反转录定量实时 PCR(qRT-PCR)检查表达变化,通过 Western blot 和免疫染色分析蛋白质,在静态孵育中定量胰岛素分泌。
NICC 成熟伴随着葡萄糖反应性胰岛素分泌的增加(刺激两倍),这是由控制β细胞身份和功能的基因的 mRNA 上调支持的,如 NEUROD1、UCN3、ABCC8 和 CASR(Log 倍数变化 [LogFC] > 1.6)。活跃的 TGFβ 受体(TGFBR)-SMAD2/3 途径促进 NICC 成熟,因为 TGFBR 抑制剂 SB431542 逆转了上述基因的上调,并解除了成熟β细胞中不允许的 ALDOB 的表达抑制。在胎球蛋白-A 处理的 NICCs 中,β细胞标志物的上调和葡萄糖反应性的开始受到抑制。同时,SMAD2/3 磷酸化受到抑制。转录组分析证实了胎球蛋白-A 和 SB431542 对 TGFβ-1 和 SMAD2/3 调节转录的抑制作用。然而,与 SB431542 相反,并且不管 cMYC 的上调,胎球蛋白-A 抑制β细胞增殖(0.27 ± 0.08% vs 控制 NICCs 中 1.0 ± 0.1% Ki67 阳性细胞)。这种效应是通过降低 FOXM1、CENPA、CDK1 或 TOP2A 的表达(LogFC ≤ -2.4)来维持的。同样,胎球蛋白-A 处理的 NICCs 中胰岛素阳性细胞的数量低于对照组 NICCs(分别为 14.4 ± 1.2%和 22.3 ± 1.1%)。在成人胰岛中,胎球蛋白-A 消除了葡萄糖反应性,即对照和胎球蛋白-A 培养的胰岛中 2.8 mmol/l 葡萄糖的 1.7-和 1.1 倍变化。此外,胎球蛋白-A 降低了 SMAD2/3 磷酸化,并抑制了增殖基因的表达。值得注意的是,在非糖尿病患者中,血浆胎球蛋白-A 与胰岛β细胞面积呈负相关(p = 0.013)。
结论/解释:我们的结果表明,围产期胎球蛋白-A 的下降减轻了胰岛中的 TGFBR 信号,这一过程促进了新生β细胞的功能成熟。功能性成熟在以后的生活中仍然可以恢复,代谢不健康的环境,如脂肪肝和升高的血浆胎球蛋白-A 的发生,可以损害β细胞的功能和适应性增殖。
本研究中产生的 RNAseq 数据集和计算机代码可在基因表达综合数据库(GEO)中获得:GSE144950;https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE144950。