Division of Biochemistry and Structural Biology, Department of Chemistry, Lund University, Lund SE-22100, Sweden.
Bioinformatics. 2021 Sep 29;37(18):2874-2881. doi: 10.1093/bioinformatics/btab205.
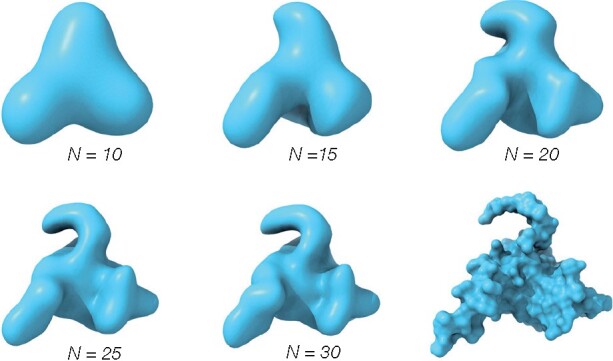
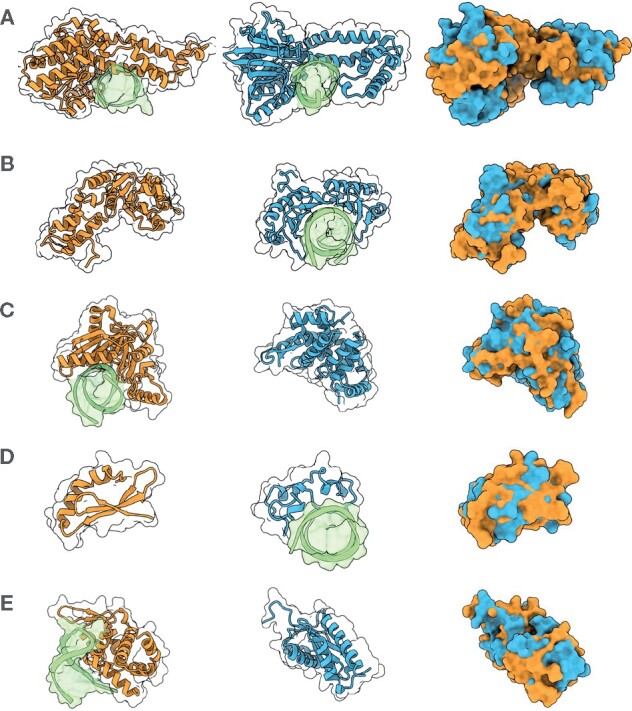
Most protein-structure superimposition tools consider only Cartesian coordinates. Yet, much of biology happens on the surface of proteins, which is why proteins with shared ancestry and similar function often have comparable surface shapes. Superposition of proteins based on surface shape can enable comparison of highly divergent proteins, identify convergent evolution and enable detailed comparison of surface features and binding sites.
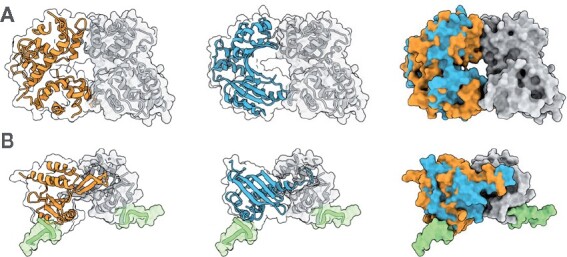
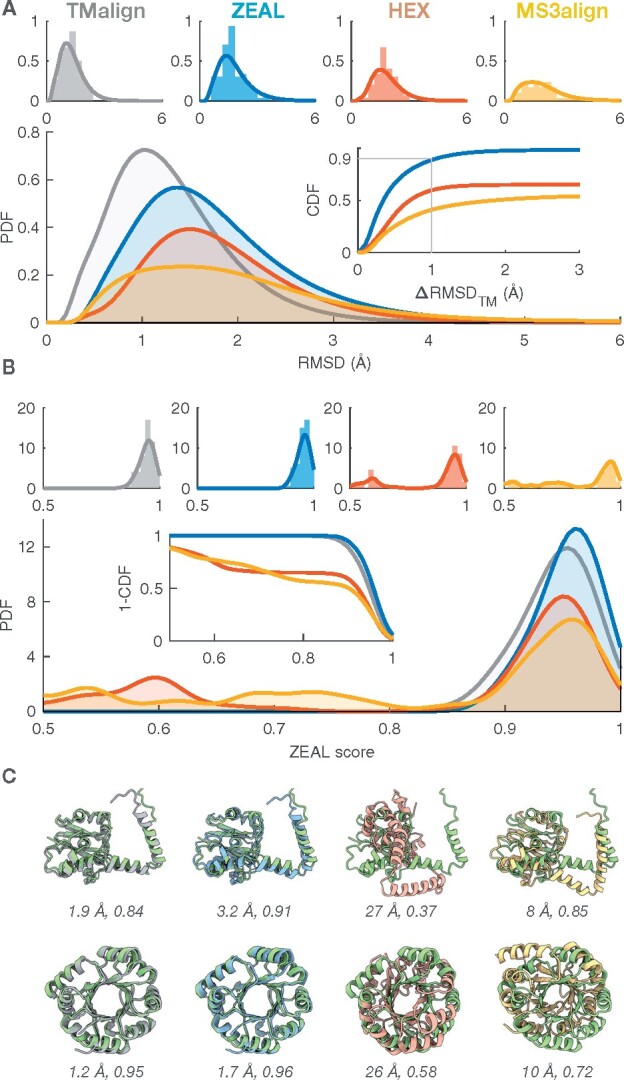
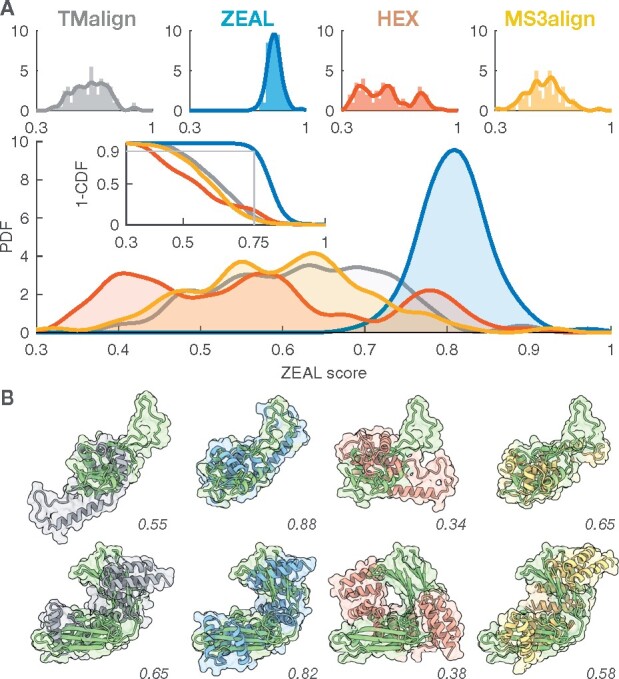
We present ZEAL, an interactive tool to superpose global and local protein structures based on their shape resemblance using 3D (Zernike-Canterakis) functions to represent the molecular surface. In a benchmark study of structures with the same fold, we show that ZEAL outperforms two other methods for shape-based superposition. In addition, alignments from ZEAL were of comparable quality to the coordinate-based superpositions provided by TM-align. For comparisons of proteins with limited sequence and backbone-fold similarity, where coordinate-based methods typically fail, ZEAL can often find alignments with substantial surface-shape correspondence. In combination with shape-based matching, ZEAL can be used as a general tool to study relationships between shape and protein function. We identify several categories of protein functions where global shape similarity is significantly more likely than expected by random chance, when comparing proteins with little similarity on the fold level. In particular, we find that global surface shape similarity is particular common among DNA binding proteins.
ZEAL can be used online at https://andrelab.org/zeal or as a standalone program with command line or graphical user interface. Source files and installers are available at https://github.com/Andre-lab/ZEAL.
Supplementary data are available at Bioinformatics online.
大多数蛋白质结构叠加工具仅考虑笛卡尔坐标。然而,生物学的很大一部分发生在蛋白质的表面,这就是为什么具有共同祖先和相似功能的蛋白质通常具有可比的表面形状。基于表面形状的蛋白质叠加可以实现高度不同的蛋白质的比较,识别趋同进化,并能够详细比较表面特征和结合位点。
我们提出了 ZEAL,这是一种交互式工具,可使用 3D(Zernike-Canterakis)函数根据形状相似性对全局和局部蛋白质结构进行叠加,以表示分子表面。在对具有相同折叠结构的结构进行的基准研究中,我们表明 ZEAL 在基于形状的叠加方面优于另外两种方法。此外,ZEAL 的对齐与 TM-align 提供的基于坐标的叠加具有可比的质量。对于序列和骨干折叠相似性有限的蛋白质比较,基于坐标的方法通常失败,而 ZEAL 通常可以找到具有大量表面形状对应关系的对齐。结合基于形状的匹配,ZEAL 可用作研究形状与蛋白质功能之间关系的通用工具。我们确定了几个蛋白质功能类别,其中在比较折叠水平上相似性较小的蛋白质时,全局形状相似性比随机机会更有可能。特别是,我们发现全局表面形状相似性在 DNA 结合蛋白中特别常见。
ZEAL 可在 https://andrelab.org/zeal 上在线使用,也可作为带有命令行或图形用户界面的独立程序使用。源文件和安装程序可在 https://github.com/Andre-lab/ZEAL 上获得。
补充数据可在生物信息学在线获得。